
As primary eye care clinicians in fast-paced clinics, it is imperative to identify whether incidental findings of foveal hypoplasia are innocuous or require further investigations. In a scenario where a clinician does identify foveal hypoplasia on optical coherence tomography (OCT), should there be changes in clinical examinations, management, and suspicions of systemic conditions?
This article delves into these topics to provide further details and practical clinical pearls.
Foveal hypoplasia is a retinal condition denoting lack of full morphological development of the fovea.1 Often, foveal hypoplasia is associated with poor visual acuity, nystagmus and low vision. However, increasing use of OCT in optometric practices2 has led to foveal hypoplasia being detected incidentally in otherwise healthy patients with no particular visual concerns.
HOW TO DIAGNOSE FOVEAL HYPOPLASIA
Traditionally, the diagnosis of foveal hypoplasia has primarily been based on funduscopic examination. Characteristic fundus appearance includes absence or irregular foveal reflex due to lack of foveal pit and, in some cases, irregular macular pigmentation. There may also be irregular distribution of peri foveal capillary vessels, with an absent foveal avascular zone, as observed with fl uorescein angiography.3
Recently, OCT has proven to be a more sensitive tool in diagnosing foveal hypoplasia in ophthalmic and optometric practices.4 Importantly, milder grades of foveal hypoplasia can present with a shallow or nearly normal foveal pit.5 Hence, lack of a foveal pit is no longer considered pathognomonic of foveal hypoplasia and instead, clinicians should inspect the continuation of inner retinal layers over the foveal region (Figure 1, red arrows).
Foveal hypoplasia presents with continuation of inner retinal layers…

Figure 1. OCT scans taken through the fovea of a healthy individual displaying A. discontinuation of inner retinal layers (yellow arrows), B. foveal hypoplasia with preserved foveal pit, and C. foveal hypoplasia with absent foveal pit. Note the continuation of inner retinal layers in (B,C) marked with red arrows.
DEVELOPMENT OF FOVEAL HYPOPLASIA
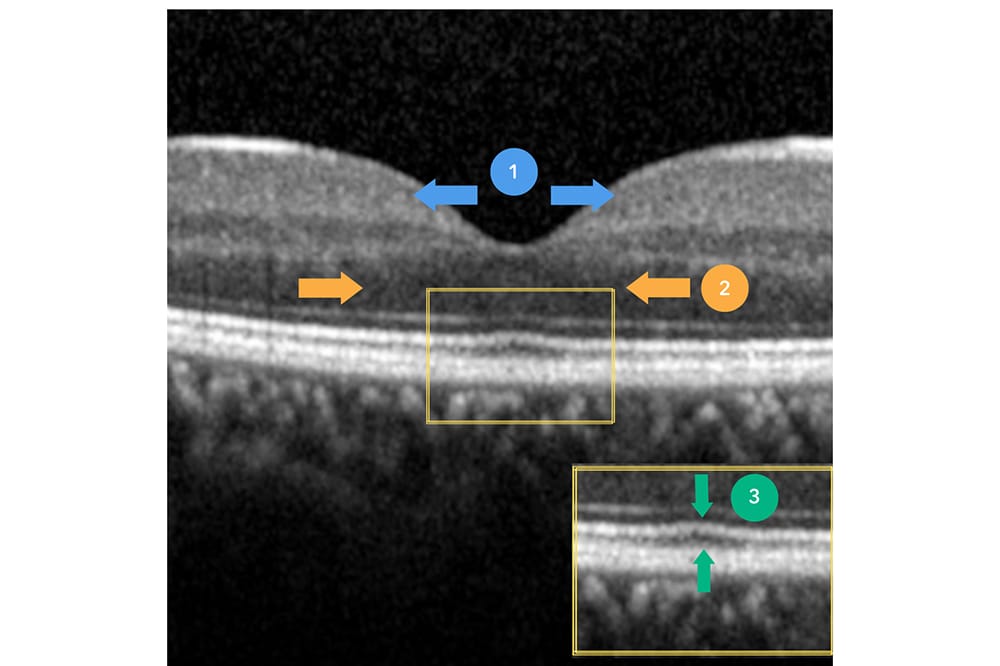
Foveal hypoplasia denotes termination (at varying stages) of normal foveal development. To understand its pathophysiology, a summary of normal foveal maturation from 11 weeks’ gestation to six-years-of-age is provided as follows:
- Inner retinal cells are centrifugally displaced towards the periphery, forming the beginning of a foveal pit (Figure 2, section 1, blue arrows). The complete extrusion of inner retinal layers occurs 15 months after birth and defines the foveal pit.
- Cone photoreceptors undergo centripetal migration towards the incipient fovea, ensuring the highest density of photoreceptors in this area. This is represented by widening of the outer nuclear layer (ONL) on OCT (Figure 2, section 2, orange arrows).
- Cone specialisation, whereby cone outer segments are lengthened (Figure 2, section 3, green arrows) and diameters are reduced to maximise cone packing density.
Foveal formation can be hindered at any point during these developmental processes, leading to various degrees of foveal hypoplasia.
DIFFERENT GRADES OF FOVEAL HYPOPLASIA
Foveal hypoplasia can be subdivided into typical and atypical forms. We will focus on typical hypoplasia, as this is most commonly found incidentally in primary care clinics.
Typical foveal hypoplasia presents with continuation of the inner retinal layers and can be associated with lack of a foveal pit; outer segment lengthening; and/or outer nuclear layer widening. The most widely accepted OCT-based classification scheme, i.e., the Leicester Grading System, suggests that higher grades of foveal hypoplasia correspond with a higher number of the aforementioned features lacking on OCT.5
However, translatability of this grading system into clinical and patient relevant outcomes is still poorly understood. Conflictingly thus far, research has shown that foveal morphology may5,6 or may not7,8 correlate with visual acuity. These studies bring into question the prognostic role of foveal morphology and further research is required to understand the complex interrelating factors that may predict clinical and patient relevant outcomes, such as visual acuity.
DIAGNOSIS OF COMMONLY ASSOCIATED CONDITIONS
A diagnosis of typical foveal hypoplasia is important in a clinical practice as it may lead to diagnosing associated conditions such as albinism, aniridia, stickler syndrome and retinopathy of prematurity.4,6 The following summary provides a brief clinical guide to differentiate potential associations of typical foveal hypoplasia.
Albinism
Albinism, the condition most commonly associated with foveal hypoplasia, is a heterogeneous genetic disorder with impaired melanin synthesis or melanin transport in the skin, hair, and eyes. There have been up to 20 genes and several subtypes of oculo-cutaneous albinism identified, with variable degrees of cutaneous hypopigmentation and ocular involvement. While the obvious cases of albinism are not challenging to diagnose, milder forms may require a thorough examination and history taking.
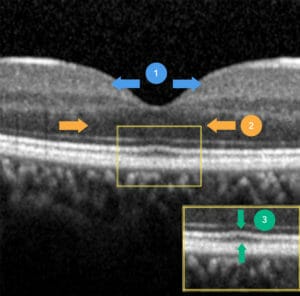
Figure 2. Critical steps involved in healthy foveal development. 1. Extrusion of inner retinal layers and pit formation, 2. centripetal migration of cone photoreceptors causing widening of outer nuclear layer, and 3.
cone specialisation displaying lengthening of cone outer segment.
An ocular-involving case is seen in Figure 3, whereby foveal hypoplasia was diagnosed in an asymptomatic patient with good visual acuity, leading to suspicions of ocular albinism.
Clinicians should be aware of nystagmus, iris translucency, and fundus hypopigmentation.9 Careful observation of skin and hair pigmentation should be made, and patients can be asked how these signs compare to their siblings and parents. These patients often have an abnormal pattern of decussation at the optic chiasm, known as chiasmal misrouting, which can be detected by visual evoked potential (VEP) testing.9
The diagnosis and appropriate referral for these patients may be critical as some syndromic forms of albinism, such as Hermansky Pudlak syndrome and Chediake Higashi syndrome, are associated with blood platelet dysfunctions and immune deficiencies,6 leading to shorter life expectancy, particularly in paediatric patients.
Aniridia/PAX6 Mutations
Aniridia is a rare congenital pan-ocular disorder often associated with PAX6 gene mutation. It is defined as complete or partial absence of the iris, and frequently associated with developmental abnormalities of the cornea, lens, optic nerve and fovea hypoplasia. While advanced cases of aniridia are often diagnosed in infancy due to complete absence of the iris, those with milder phenotypes and more subtle signs may require the observation of a meticulous clinician. Signs are partial absence of the iris or milder iris defects, such as pseudocoloboma, eccentric pupil, corectopia, or ectropion uveae.10
The diagnosis of aniridia can be significant, as these patients are at higher risk of developing glaucoma, aniridia associated keratopathy, cataract formation and dry eye disease, and hence ongoing monitoring may be required.11

Figure 3. A 21-year-old asymptomatic female was referred to the Centre for Eye Health for assessment of optic nerve head. Her unaided visual acuities were 6/7.5+ right and 6/7.5+ left. A. While optic nerve head findings were unremarkable, incidental findings included mottled macular pigmentation and fundus hypopigmentation. B. OCT scans revealed continuation of inner retinal layers and lack of outer segment lengthening over the presumed foveal region. C. Close inspection of the anterior
segment revealed bilateral transillumination defects. Upon further questioning, she noted lighter skin and hair colour compared to her siblings, most consistent with a diagnosis of albinism. She was referred for assessment of visual evoked potential for confirmation of diagnosis.
Stickler Syndrome
Stickler syndrome is an inherited connective tissue disease affecting multiple organs including eyes, ears, joints, and midline facial structures. It has been shown that up to 82% of eyes with stickler syndrome present with foveal hypoplasia.12 The clinical presentation may vary widely, however some common features include midface hypoplasia, hearing loss of variable severity, arthritis, and joint hyper-mobility along with ocular signs of high myopia, vitreous abnormalities, retinal detachments, and congenital cataracts.
Affected individuals are at significant risk of recurrent retinal detachments and glaucoma development. Through early detection, visual outcomes can be improved for these patients.12
Retinopathy of Prematurity
Retinopathy of prematurity (ROP) is a vaso-proliferative disease affecting preterm or low birthweight infants. It is characterised by avascularised peripheral retina and subsequent cicatricial vitreoretinal changes. There is a high association between patients with a history of ROP and foveal hypoplasia, with up to 91% of patients presenting with hypoplasia.13 Clinicians should ask thorough questions regarding birth history, in conjunction with dilated fundus examinations, observing for signs of peripheral vascular straightening, pigmentary changes, cicatricial vitreoretinal interface abnormalities, peripheral retinal folds, lattice degenerations, retinal schisis, as well as signs of tractional and rhegmatogenous retinal detachments.14 These patients require ongoing monitoring for complications developing in adulthood, such as early cataract formation, early onset glaucoma, high myopia, and high risk of retinal tears and breaks.
Idiopathic Foveal Hypoplasia
While some presentations of foveal hypoplasia are associated with systemic conditions, it can also be an isolated and idiopathic finding in an asymptomatic patient.8,15 This diagnosis should be made through systematically excluding associated conditions mentioned above. Although idiopathic foveal hypoplasia may be associated with some genetic mutations, the genetic research is complex and still ongoing in identifying genotypic and phenotypic spectrum of foveal hypoplasia.6 Patients with otherwise unremarkable ocular findings would need no further examinations and can be monitored with routine optometric examinations.

Table 1. Summary of clinical features and management for commonly associated conditions with foveal hypoplasia.
SUMMARY
In clinical practice, the use of OCT has been profound in detecting foveal hypoplasia, particularly in asymptomatic patients. Continuation of inner retinal layers on OCT scan is a pathognomic sign, which can also be accompanied by lack of foveal pit, outer segment lengthening and/or outer nuclear layer widening. Clinicians should be aware of commonly associated conditions such as albinism, aniridia, stickler syndrome and ROP and, with thorough examination, exclude these prior to making a diagnosis of idiopathic foveal hypoplasia.
Meri Galoyan, BOptom(Hons) UNSW is a staff optometrist at the Centre for Eye Health. Through her work at the Centre, she has developed strong interest in advanced ocular imaging and macular diseases. She enjoys applying her knowledge to provide evidence-based care for her patients. Ms Galoyan is actively involved in clinical services as well as student supervision and teaching.
The author thanks Pauline Xu and Matt Trinh for reviewing the manuscript.
The Centre for Eye Health receives primary funding from Guide Dogs NSW/ACT.
References
- Thomas MG, Papageorgiou E, Kuht HJ, Gottlob I. Normal and abnormal foveal development. British Journal of Ophthalmology. 2020:bjophthalmol-2020-316348.
- Le HM, Souied EH, Pedinielli A, Zambrowski O, Miere A. Idiopathic foveal hypoplasia: quantitative analysis using optical coherence tomography angiography. Retina (Philadelphia, Pa). 2020;40(12):2325-31.
- Batıoğlu F, Demirel S, Özmert E, Bayraktutar B, Yanık Ö. The diagnostic role of multimodal imaging techniques in isolated foveal hypoplasia. Turk J Ophthalmol. 2017;47(5):306-8.
- Kondo H. Foveal hypoplasia and optical coherence tomographic imaging. Taiwan journal of ophthalmology. 2018;8(4):181-8.
- Thomas MG, Kumar A, Mohammad S, Proudlock FA, Engle EC, Andrews C, et al. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology. 2011;118(8):1653-60.
- Kuht HJ, Maconachie GDE, Han J, Kessel L, van Genderen MM, McLean RJ, et al. Genotypic and phenotypic spectrum of foveal hypoplasia: a multicenter study. Ophthalmology. 2022;129(6):708-18.
- Mota A, Fonseca S, Carneiro A, Magalhães A, Brandão E, Falcão-Reis F. Isolated foveal hypoplasia: tomographic, angiographic and autofl uorescence patterns. Case reports in ophthalmological medicine. 2012;2012:864958.
- Kirchner ID, Waldman CW, Sunness JS. A series of fi ve patients with foveal hypoplasia demonstrating good visual acuity Retinal cases & brief reports. 2019;13(4):376-80.
- Kruijt CC, de Wit GC, Bergen AA, Florijn RJ, Schalij- Delfos NE, van Genderen MM. The phenotypic spectrum of albinism. Ophthalmology. 2018;125(12):1953-60.
- Daruich A, Robert MP, Leroy C, N DEV, Beugnet C, Malan V, et al. Foveal hypoplasia grading in 95 cases of congenital aniridia: correlation to phenotype and PAX6 genotype. Am J Ophthalmol. 2022;237:122-9.
- Landsend ECS, Lagali N, Utheim TP. Congenital aniridia – A comprehensive review of clinical features and therapeutic approaches. Survey of Ophthalmology. 2021;66(6):1031-50.
- Matsushita I, Nagata T, Hayashi T, Kimoto K, Kubota T, Ohji M, et al. Foveal hypoplasia in patients with stickler syndrome. Ophthalmology. 2017;124(6):896-902.
- Villegas VM, Capó H, Cavuoto K, McKeown CA, Berrocal AM. Foveal structure-function correlation in children with history of retinopathy of prematurity. Am J Ophthalmol. 2014;158(3):508-12.e2.
- Kaiser RS, Trese MT, Williams GA, Cox MS, Jr. Adult retinopathy of prematurity: outcomes of rhegmatogenous retinal detachments and retinal tears. Ophthalmology. 2001;108(9):1647-53.
- Giocanti-Aurégan A, Witmer MT, Radcliffe NM, D’Amico DJ. Isolated foveal hypoplasia without nystagmus. Int Ophthalmol. 2014;34(4):877-80.





