Assessing the optic disc in a child is challenging, but critical for the paediatric ophthalmology assessment. Presented here are common optic disc anomalies seen in clinical practice, with tips for assessment, investigation and diagnosis.
CLINICAL ASSESSMENT
As with all components of a paediatric examination, the techniques and strategies used to acquire the necessary information will be determined by the child’s age, mood, interest and their ability to cooperate on that day. Many children are able to co-operate for slit-lamp biomicroscopy from approximately five years of age. For younger children, those with autism or developmental challenges, or those who remain fearful despite reassurance, slit lamp biomicroscopy is not appropriate. The most valuable tool to examine the optic disc in children, and the one I use as first line in every patient, is a binocular indirect ophthalmoscope and a 28D lens (or 20D lens, or equivalent if higher magnification is required). The child’s pupils should be dilated, the room lights dimmed, and the examination light lowered (to facilitate cooperation). I ask the child to look at my ear or draw their attention there with my fingers or a small toy. With regards to an optic disc examination, I make an assessment on the clarity of the disc margin and height of the overlying nerve fibre layer, the disc size, the cup-to-disc ratio, the presence or absence of any disc pallor or pigmentary anomalies, the posterior pole vasculature, the presence or absence of any haemorrhages or cotton-wool spots, the macula and its light reflex, and the retinal periphery. Where indicated, I also look for spontaneous venous pulsations and if they are not present, I will apply gentle pressure on the globe and see if they are easily induced.
For younger children, those with autism or developmental challenges, or those who remain fearful despite reassurance, slit lamp biomicroscopy is not appropriate

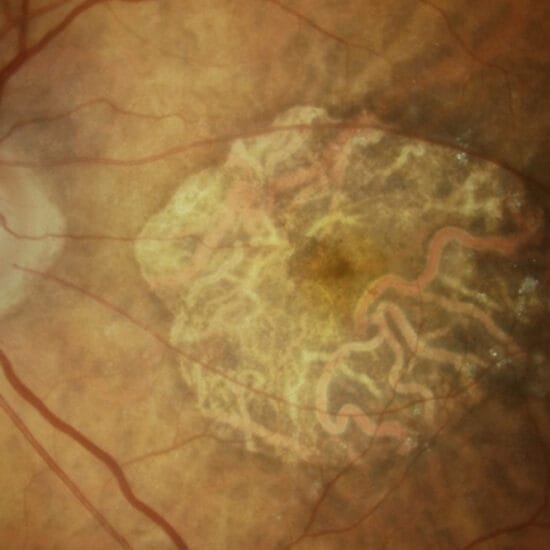
Figure 1. Optic disc drusen in a 10-year old boy. This patient was referred after his optometrist noted bilateral disc elevation, more prominent in the left eye (B) than the right (A), an incidental finding at routine eye review. Each disc is elevated and the margins are blurred. There is no obscuration of disc margin vessels and spontaneous venous pulsations are present bilaterally. There is vessel trifurcation at the disc on the left (B). There are areas of autoflourescence in the inferior quadrant of the right eye (C) and left eye (D) discs, supporting the diagnosis of optic disc drusen. An EDI-OCT scan through the right disc drusen is shown in Figure 3.
It is important to attempt a complete assessment in every child wherever possible because clinical findings lend support to each other and it is difficult (and dangerous) to make decisions based on an isolated clinical sign. The most important additional components of the assessment, in the context of optic disc anomalies, are: monocular visual acuities; pupil responses to light; ocular alignment and motility; and refraction after cycloplegia.
Fundus images are powerful tools for assessing the disc as they give a static image that can be reviewed without time pressure, and they provide a baseline for future comparison. Wide-field digital imaging systems, such as the Optos PLC Optomap, can be used in very young children, and even babies. The RetCam3 is appropriate for neonates or children being examined under general anaesthesia. Standard fundus photography and optical coherence tomography (OCT) is often possible beyond the age of approximately five years. Automated perimetry is challenging for children and unlikely to be reliable under the age of approximately 12 years. Kinetic perimetry (such as the Goldman visual field test) is much more reliable but is not widely available and it requires a highly skilled operator to perform the test. Visual fields can be assessed clinically, using a confrontation technique. B-scan ultrasonography is a helpful investigation, particularly in the context of confirming the presence of disc drusen. It is quite well tolerated and can give results in children of all ages. Enhanced Depth Imaging (EDI)-OCT is being used with increasing frequency to image the disc to the level of the lamina cribrosa.
PAPILLOEDEMA
This condition is defined as swelling or oedema of the optic nerves, secondary to elevated intracranial pressure. Profound and irreversible vision loss may occur as a result of optic nerve damage. Raised intracranial pressure may be idiopathic (as in idiopathic intracranial hypertension) or secondary to other causes (such as a brain tumour or structural brain anomaly). The diagnosis of papilloedema is made by identifying optic disc oedema in combination with elevated intracranial pressure measured by lumbar puncture or directly (intracranially).1 Systemic features of raised intracranial pressure in children include:2
- Headache,
- Worse at night or upon awakening,
- Associated with transient visualobscuration on postural changes,
- Associated with nausea and vomiting,
- Constitutional symptoms,
- Poor feeding,
- Mood change,
- Lethargy,
- Failure to thrive, and/or
- Bulging fontanelles or rapid increasein head circumference.
Clinical features suggestive of papilloedema include disc margin blur and elevation with obscuration of disc margin blood vessels; venous engorgement; loss of spontaneous venous pulsations or difficulty inducing them; the presence of haemorrhages; and symptoms associated with raised intracranial pressure. It is important to note though, disc oedema may be quite mild and can be difficult to distinguish from pseudo-papilloedema, such as buried disc drusen or congenitally crowded discs. If a child is suspected of having papilloedema, an urgent referral should be made, for a full ophthalmic assessment and neuroimaging of the form of MRI/MRV. Lumbar puncture can be used to confirm elevated intracranial pressure, where indicated.

Figure 2. B scan ultrasound of suspected disc drusen in an eight-year old girl. This child was referred by her ophthalmologist for a second opinion. She was initially seen by her optometrist who noted bilateral disc margin blur and elevation at routine eye examination. She underwent an extensive workup, including neuroimaging and lumbar puncture, which were normal. The B scan ultrasound (A) shows hyperechoic foci in each optic nerve head (arrows) without posterior acoustic shadowing. This appearance may indicate nonmineralised drusen. The colour fundus photographs of her optic discs show vessel trifurcation at the right (B) and left (C) discs. There is no obscuration of the disc margin vessels and venous pulsations were easily induced.
PSEUDOPAPILLOEDEMA (OPTIC DISC DRUSEN)
This is a challenging diagnosis to make in young children because drusen are often buried deep within the nerve head and are not yet calcified. This means the disc appears elevated with a blurred margin but does not necessarily have the characteristics we typically associate with drusen. This creates a diagnostic dilemma as it can be very challenging to distinguish drusen from papilloedema in young children.
Drusen are often an incidental finding and are rarely symptomatic. They may, however, be associated with transient visual obscurations and visual field defects. Visual field defects are more common in superficial drusen and progress very slowly with advancing age.3 The clinical features of disc drusen include optic disc elevation and ‘lumpiness’, crowding with no significant cup, disc pallor, and vascular branching anomalies (particularly vessel trifurcation). There may be an associated cilioretinal artery.4 Fundus autofluorescence (Figure 1) can help in diagnosis but is more sensitive if the drusen are superficial rather than buried. B scan ultrasound shows drusen as hyperechoic spots within the nerve head (Figure 2), with posterior shadowing, and has long been considered the gold standard test for diagnosing disc drusen. This characteristic appearance only occurs once the drusen calcify, which occurs at a mean age of 8.8 years.3 EDI-OCT is showing much promise in its ability to image the disc to the level of the lamina cribrosa (Figure 3) and diagnose disc pathology. From a study of patients over the age of 18 years without other pathology, the Optic Disc Drusen Studies Consortium5 defines optic nerve head drusen as structures on EDI-OCT with the following characteristics:
- Located above the lamina cribrosa,
- Signal poor cores,
- Often with a hyperreflective marginat the superior edge,
- They are distinct from peripapillaryhyperreflective ovoid mass-like structures,and
- Hyperreflective horizontal lines mayrepresent early optic disc drusen.Other causes of pseudopapilloedema include tilted discs and disc crowding in the context of high hypermetropia.
CONGENITAL OPTIC DISC ANOMALIES
Congenital optic disc anomalies may be unilateral or bilateral. Bilateral anomalies may present in early childhood with reduced visual responses and nystagmus. Unilateral anomalies may present later with amblyopia and sensory strabismus or be found incidentally. A trial of occlusion therapy for treatment of amblyopia is always warranted where the structural anomaly and vision is asymmetrical. Congenital optic disc anomalies are frequently associated with other central nervous system malformations.

Figure 3. EDI-OCT of optic disc drusen in a 10-year old boy. This is the same patient as in Figure 1, with the raster taken through the area where autofluorescence indicates a druse is present. The thin white arrow indicates a signal-poor core. The thick white arrows indicate a hyperreflective margin. The thin yellow arrow indicates a horizontal hyperreflective line. These features are supportive of the diagnosis of optic disc drusen.5
Optic Nerve Hypoplasia
Optic nerve hypoplasia is the most common type of congenital optic disc anomaly seen in clinical practice. The optic disc is small, grey or pale, and is often surrounded by a ring of depigmentation (the ‘double ring’ sign). The outer ring, which represents the scleral canal, can be misinterpreted as the outer edge of the optic nerve leading to a delay in diagnosis. The disc vessels may be tortuous. The visual acuity can be normal or as poor as no light perception. Measuring the ratio of the horizontal disc diameter to the disc-macula distance may predict visual acuity outcomes: greater than 0.3 is associated with good visual acuity; less than 0.3 is associated with reduced visual acuity; and less than or equal to 0.15 is associated with visual acuity of light perception or worse.6
Optic nerve hypoplasia is often associated with central nervous system abnormalities such as septo-optic dysplasia. There are also systemic associations (including albinism and aniridia) and perinatal risk factors (including fetal alcohol syndrome and maternal use of illicit drugs in pregnancy).2 The condition is thought to arise from a primary failure of retinal ganglion cell differentiation in the fourth to sixth week of gestation and a deficiency of molecules at the optic disc to guide axons. Management involves diagnosing and managing associated systemic disease (such as pituitary hormone deficiencies), correcting refractive error (there is a strong association with astigmatism) and amblyopia therapy where appropriate.
Excavated Disc Anomalies
In each of these conditions, there is an excavation of the posterior globe which surrounds and incorporates the optic disc. Each diagnosis is a distinct clinical entity, with a specific embryological origin.
Morning Glory Disc Anomaly
So-named because it resembles the morning glory flower, the optic disc is incorporated within the funnel-shaped excavation. The disc is often enlarged with a wide ring of chorioretinal pigmentary disturbance, a central tuft of glial tissue and its vessels arise from its outer edge, increased in number and straighter than the usual course. This is frequently unilateral and vision is often poor (6/60 to count fingers), though there are cases with normal vision. This condition may be associated with trans-sphenoidal basal encephalocoele (in which a meningeal pouch containing intracranial contents protrudes through a defect in the sphenoid bone) and facial characteristics including a flattened nasal bridge, hypertelorism and a notch in the middle of the upper lip. It may also be associated with hypoplasia of ipsilateral intracranial vasculature. The embryological origins are disputed but are likely related to a primary mesenchymal abnormality.
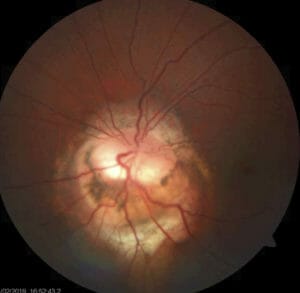
Figure 4. Contractile peripapillary staphyloma in a nine-year old girl. This child was referred for a second opinion of her left optic disc appearance. The right optic disc was normal. She gave a history of daily episodes of transient loss of vision in the left eye, comprising a complete black out of her vision “like my eye is closed”. She had vision of 6/6 in each eye, a left relative afferent pupillary defect and a very enlarged blind spot on visual field testing. The left disc was at the bottom of a deep excavation, which had straight (not funnel shaped) walls. The disc was entirely within the excavation, with vessels arising from its centre, and there was peripapillary atrophy and pigment disturbance. During the examination, the disc moved anteriorly within the excavation and then anteriorly and posteriorly in a pulsatile manner. The surrounding retinal vasculature became tortuous and began ‘blinking’ in time with the movement, presumably in time with the patient’s arterial pulse. During the episode, the patient experienced a complete blackout of her vision, consistent with her previous episodes. Her vision, and the baseline appearance of her disc returned to normal within 30 seconds.
Peripapillary Staphyloma
This is extremely rare and usually unilateral. A relatively normal-looking disc sits in the base of a deep fundus excavation. The walls of the excavation may show pigmentary change and atrophy of the RPE and choroid. In contrast to the morning glory anomaly, the disc and its vessels are relatively normal in appearance and there is no central glial tuft. The wall may contain contractile elements which cause transient visual obscurations (Figure 4). The embryological origins are likely related to incomplete differentiation of the posterior sclera with herniation of ocular tissues through the defect.
Optic Disc Coloboma
In contrast to the first two types of disc anomalies, where the disc lies within the excavation, in optic disc coloboma the excavation lies within the disc. The coloboma is a sharply demarcated, white excavation involving the inferior portion of the disc and its rim, with relative sparing of the superior rim. This inferior displacement reflects the embryological origins and defective closure of the embryonic fissure. It is frequently bilateral and the effect on visual acuity is difficult to predict based on disc appearance. They may be sporadic or autosomal dominant. They may be associated with systemic anomalies such as CHARGE syndrome.
HEREDITARY AND TOXIC-METABOLIC OPTIC DISC ANOMALIES
Primarily the result of mitochondrial dysfunction due to an exogenous agent (toxic), deficient substrate (metabolic and nutritional) or abnormal mitochondrial protein products (due to an underlying genetic defect), the visual dysfunction in these conditions is typically bilateral, symmetrical and characterised by dyschromatopsia, loss of visual acuity and field.7 The visual field loss is typically centroceacal and reflects injury to the papillomacular nerve fibre bundle, located between the fovea and the optic disc. Taking a comprehensive medical and family history are crucial in determining an underlying cause in these cases.

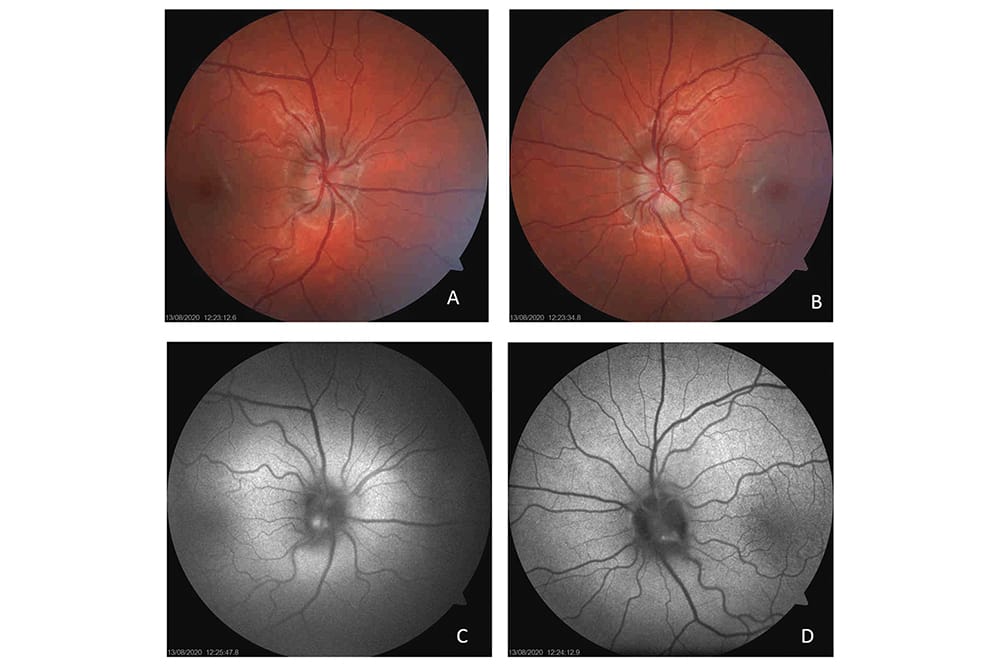
Figure 5. Dominant optic atrophy in a 10-year old boy. This boy’s mother and maternal grandfather are also known to have dominant optic atrophy. Both right (A) and left (B) optic discs show prominent temporal pallor.
Toxic and Metabolic Optic Neuropathies
Some antibiotics (particularly the anti-tuberculosis agent ethambutol), immunomodulators (such as infliximab) and chemotherapeutics (such as vincristine) can cause mitochondrial dysfunction and an optic neuropathy. Vigabatrin (an anti-epileptic agent used in the treatment of infantile spasms) causes a retinal toxicity with an atypical secondary optic nerve atrophy which spares the temporal quadrant of the disc.7 Exposure to these agents may trigger or exacerbate vision loss in individuals with an underlying genetic defect such as Lebers optic neuropathy. Nutritional optic neuropathies may affect children who are malnourished or have severely restricted and stereotyped diets. This is sometimes seen in children with autism spectrum disorder with a strong aversion to foods of a particular colour or texture, or who will eat only one food type, such as white rice or potato chips. It can rarely be associated with anorexia nervosa.2 The commonest nutritional deficiencies which can cause an optic neuropathy are vitamin B12 (Cobalamin), folate and copper.
Lebers Hereditary Optic Neuropathy
More than 90% of all Lebers hereditary optic neuropathy cases are attributable to one of three mutations in different nicotinamide adenine dinucleotide dehydrogenase (NADH) genes located within the mitochondrial deoxyribonucleic acid (DNA).7 The condition typically presents with sudden onset unilateral painless vision central loss, with fellow eye involvement within a few weeks. The peak age of onset is within the second and third decades of life. There is incomplete penetrance of the condition, with male carriers much more likely to become symptomatic (50% lifetime risk) compared with females (10% lifetime risk) even with the same mutation. At presentation, the disc may be completely normal or slightly hyperemic, with mild disc swelling and tortuosity of the central retinal vessels. Pathological cupping may develop in time. Lebers hereditary optic neuropathy plus is used to describe the condition occurring in combination with systemic findings, such as cardiac arrythmias, peripheral neuropathy, dystonia and ataxia. The visual prognosis is poor, though there can be some recovery related to genetic mutation subtype, and no treatment has been proven effective to date. Gene therapy trials are being undertaken for Lebers hereditary optic neuropathy. Because mitochondrial DNA is inherited maternally (all of an individual’s mitochondria come from the egg, none from the sperm), all children of an affected mother will carry the mutation but the children of affected fathers will not.7

Figure 6. Dominant optic atrophy. Image A is a colour fundus photo of both fundi taken in 2017 when this child (the sister of the patient in Figure 5) was five years of age and the temporal disc pallor was becoming evident. Image B was taken three years later, at which stage her vision had deteriorated in each eye and bilateral temporal disc pallor was much more prominent.
Dominant Optic Atrophy
The majority of cases are attributable to a defect in the OPA1 gene on chromosome three, which encodes an inner mitochondrial membrane protein. The condition presents with insidious bilateral painless loss of vision, usually beginning in the first decade of life. The most obvious clinical feature in those with established disease is prominent temporal disc pallor, appearing as a white triangular defect in the disc (Figures 5 and 6). Being autosomal dominant, there is a 50% chance of an affected individual transmitting the gene to children. The penetrance is between 60–90% and there is variable phenotypic expression with some individuals having greater vision loss than others. Rarely, this condition can be associated with systemic features (deafness, progressive external ophthalmoplegia, myopathy, ataxia, and neuropathy) and is denoted optic atrophy plus.7
OTHER OPTIC NEUROPATHIES OF CHILDHOOD
Traumatic Optic Neuropathy
Traumatic optic neuropathy is defined as a sudden reduction in visual function following direct or indirect trauma to the optic nerve. Indirect trauma is the more common mechanism and is thought to be due to injury to small vessels, which in turn causes ischemia, inflammation, ganglion cell death and optic atrophy. In children, the commonest aetiologies are falls, traffic accidents and sporting injuries.2 The diagnosis is made on clinical history and examination findings: presenting visual acuity may be very poor and in the acute phase, the optic disc may appear normal but a relative afferent pupillary defect and suboptimal optic nerve function parameters will be evident. It is very important to exclude reversible causes, such as a retrobulbar haematoma. Treatment is controversial and no individual treatment has been proven to be better than observation alone. The visual outcome in these cases is closely related to the vision at presentation. Poor prognostic features include poor vision at presentation, no recovery within the first 48 hours and absent visual evoked potentials on electrophysiology.2

Figure 7. Left traumatic optic neuropathy in a six-year old boy. This child had an unwitnessed fall from a bike with complete loss of vision in his left eye. At presentation he had left eye vision of 6/60, a left relative afferent pupillary
defect (RAPD) but a normal disc examination. Over the next few weeks, the left vision improved to 6/12 (right 6/6), the RAPD persisted and the left disc became pale, particularly temporally (A). The OCT of the discs (B) shows thinning of the left retinal nerve fibre layer and (C) ganglion cell loss.
CONCLUSION
A challenging but critical part of every paediatric eye examination, the optic disc assessment requires clinical skill which continue to develop with practice, and sometimes fundus imaging and other investigation techniques. A comprehensive history, family history and a thorough ophthalmic assessment can help guide diagnosis.
To earn your CPD points from this article visit mieducation.com/assessing-and-diagnosing-thepaediatric- optic-disc Dr Caroline Catt MBBS, MMed, BMedSci (Hons1), FRANZCO specialises in paediatric and general ophthalmology, including cataract surgery. She graduated with degrees from the University of Sydney and the Flinders University of South Australia and completed her ophthalmology training at Sydney Eye Hospital before undertaking a fellowship at the Hospital for Sick Children in Toronto, Canada. She was the recipient of the prestigious Dr JD Morin award for excellence as a clinical fellow in the areas of clinical care, research and education.
Dr Catt has published in peer-reviewed journals, has undertaken clinical studies in paediatric ophthalmic disease and presented the findings internationally. She has a keen interest in teaching ophthalmologists-in-training and is a Clinical Senior Lecturer with the University of Sydney, Department of Ophthalmology.
Acknowledgement
The author gratefully acknowledges the support of Professor Frank Martin and Associate Professor Clare Fraser for reviewing the EDI-OCT optic disc drusen images in Figure 3.
References
- Heidary G. Pediatric Papilledema: Review and a Clinical Care Algorithm. International Ophthalmology Clinics 2018:58(4);1-9
- Hoyt CS and Taylor D. Pediatric Ophthalmology and Strabismus Fourth Edition. Saunders Elsevier, Edinburgh, 2013
- Chang M & Pineles S (2016) Optic disk drusen in children. Survey of Ophthalmology 61:745-758
- Flores-Rodriguez P, Gili P & Martin-Rios M (2012) Ophthalmic Features of Optic Disc Drusen. Ophthalmologica 228(1):59-66
- Malmqvist L, Bursztyn L, Costello F, Digre K, Fraser JA, Fraser C, Katz B, Lawlor M, Petzoid A, Sibony P, Warner J, Wegener M, Wong S and Hamann S. The Optic Disc Drusen Studies Consortium Recommendations for Diagnosis of Optic Disc Drusen Using Optical Coherence Tomography. Journal of Neuro-Ophthalmology 2018:38;299-307
- Borchert M, McCulloch D, Rotha C and Stout A. Clinical Assessment, Optic Disk Measurements and Visual-Evoked Potential in Optic Nerve Hypoplasia. American Journal of Ophthalmology 1995;120;5;605-12
- Oliveira C. Toxic-Metabolic and Hereditary Optic Neuropathies. Continuum Journal 2019:5;1265-88.