Although the majority of paediatric ophthalmic tumours are benign, prompt diagnosis and management are crucial as these lesions may affect vision, cause deformity or have systemic implications. This article reviews the epidemiology, clinical manifestations, and the neuro-imaging features of common paediatric orbital lesions.
Paediatric orbital tumours can pose a diagnostic dilemma as they are relatively uncommon and have a varied presentation in comparison to adults. A sound understanding of the disease spectrum and various ways it may manifest is crucial, particularly to those of us who see children in our practices. Paediatric orbital tumours are largely categorised as neoplastic (benign versus malignant) and vascular. Pattern recognition is useful to distinguish the early versus late onset tumours, which can significantly impact upon a child’s morbidity and mortality.
Table 1 presents a useful summary.
Pattern recognition is useful to distinguish the early versus late onset tumours, which can significantly impact upon a child’s morbidity and mortality

Table 1. A summary of paediatric orbital tumours.
BENIGN LESIONS
Dermoid and Epidermoid Inclusion Cysts
Orbital cystic lesions are the most common paediatric orbital lesions. Age of onset varies from childhood to early teenage years. They are largely benign and can be congenital or acquired. Dermoid cysts are the most common lesion, arising from ectoderm trapped in bony sutures during embryogenic migration. Epidermoid cysts present similarly, lacking dermal elements within the cyst wall.
Clinical Presentation

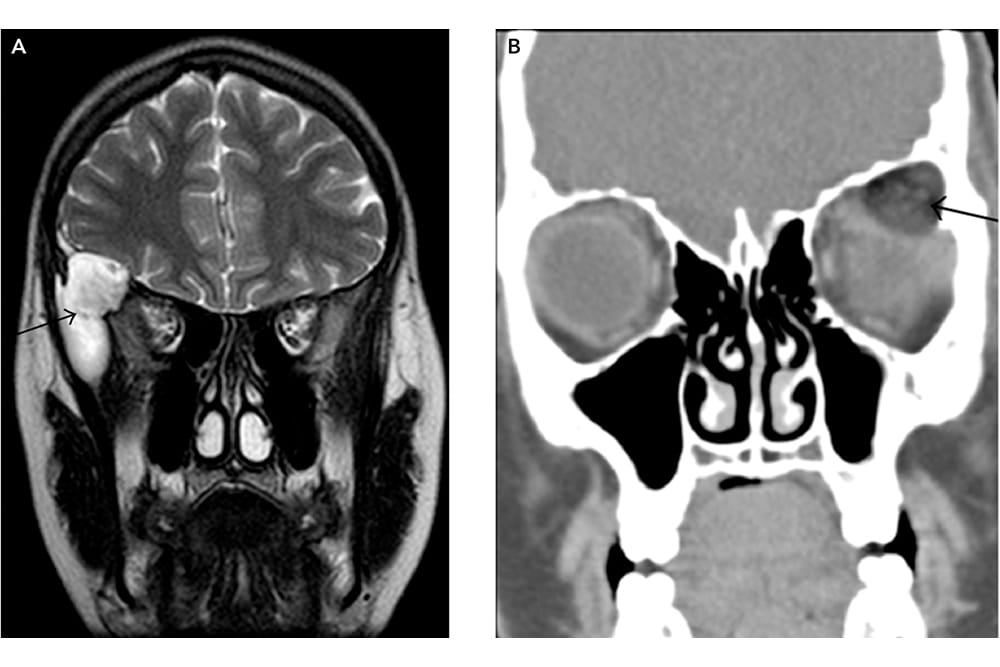
Figure 1. A: MR coronal T2-weighted image shows a hyperintense mass in the right superolateral orbit and sphenoid wing, consistent with an epidermoid inclusion cyst and B: CT coronal with soft-tissue algorithm show low attenuation fat within the mass (black arrow), diagnostic of a dermoid inclusion cyst.17
These lesions typically present as a slow growing, painless, slightly fluctuant subcutaneous mass, near the supero-temporal orbit and fronto-zygomatic suture. Globe displacement and vision reduction is rare. Deep orbital or intraconal dermoid cysts (Figure 1) may present with proptosis, ocular motility disturbances and orbital nerve compression. Dumbbell dermoids can be found anteriorly as palpable masses but extend into the orbit and further posteriorly into the temporal fossa, through destruction of the lateral orbital wall. Neuroimaging is therefore vital when assessing these lesions, given the possibility of posterior extension. Rupture, either spontaneously or secondary to trauma, results in an intense inflammatory response that may mimic orbital cellulitis.1 Additionally, orbital fistulas or sinus tracts can be contiguous with the dermoid, resulting in orbital cellulitis.2

Figure 2. A: MR axial T2-weighted image and B: T1 axial-weighted post-contrast image showing sphenoid wing dysplasia with mild temporal lobe herniation (black arrow); plexiform neurofibroma with areas of T2 hyperintensity, heterogeneous enhancement, and extension lateral to the orbit into the infratemporal fossa (curved arrow), buphthalmos and exophthalmos (star) of the right orbit.17
Neuroimaging
Ultrasound (superficial dermoid)
Smooth contours,
Variable echogenicity, and
No demonstrable internal vascularity.
MRI
Well circumscribed unilocular cysts,
Isointense on T1W1,
Hyperintense on T2W1, and High signal intensity on diffuse weighted imaging (DWI).

Figure 3. A: Coronal contrast-enhanced CT demonstrates a well-defined, osteolytic mass in the left orbital roof with rim enhancement and B: MR coronal T2-weighted fat-saturated image shows heterogeneous signal, consistent with LCH.17
CT
Dermoid and epidermoid cysts cause adjacent bony remodelling, demonstrated as an adjacent rim of dense bone.
Dermoid cysts have low attenuation fat and post-contrast enhancement of the wall.
Plexiform Neurofibroma
Plexiform neurofibroma (PNF) is a hamartoma of neuroectodermal origin, representing 1–2% of all orbital tumours, typically arising in the first decade of life.3 Presence of a PNF is diagnostic of neurofibromatosis 1 (NF1). The lesion can involve any peripheral nerve but usually involves sensory nerves in the orbit or eyelid, causing widening of the superior orbital fissure and greater sphenoid wing dysplasia4 (Figure 2).

Figure 4. A: T1WI coronal image showing the well-circumscribed, mildly hypoattenuating mass in the right lacrimal gland that homogenously enhances on B: postcontrast T1-weighted image, consistent with lymphoid hyperplasia.17
Clinical Presentation
Eyelid PNFs have a characteristic S shape and feel like ‘a bag of worms’ on palpation. Growth can result in complete ptosis, occlusion amblyopia, anisometropia, strabismus and discomfort due to rubbing of the lashes against the upper palpebral conjunctiva.4 Ipsilateral glaucoma is reported in up to 50% of patients.4
Orbital involvement can cause globe dystopia, bony expansion of the anterior orbital rim, orbital foramina and middle cranial fossa, and sphenoid dysplasia, resulting in temporal lobe herniation and pulsatile exophthalmos.4 Orbital infiltration can subsequently lead to involvement of the sensory nerves, lacrimal gland, and extraocular muscles. Risk of malignant transformation to a sarcoma is reported in up to 7–10% of cases.4
Neuroimaging

Figure 5. A: MR axial T2-weighted fat saturated and B: coronal T1-weighted fat saturated images showing an infiltrative, diffusely enhancing soft-tissue mass involving the left superior rectus muscle (black arrows), consistent with OIS.17
MRI
Diffuse, irregular masses that cross multiple tissue planes,
Thickening of the soft tissues,
Irregular intraconal fat,
Irregular nodularity of the optic nerve sheath,
Thickening of the sclera/choroid,
Demonstrates brain herniation through defects in the sphenoid,
Hypointense on T1WI,
Hyperintense on T2WI, and
Variable enhancement on postcontrast images.

Figure 6. MR axial T1-weighted post-contrast fatsaturated images demonstrate homogeneously enhancing infiltrative tissue behind the prosthesis (black arrows). Increased signal along the course of the optic
nerve is consistent with perineural spread of disease (curved white arrow), consistent with lymphoma.17
Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH, Figure 3), a disease characterised by abnormal proliferation of Langerhans cells, represents a spectrum of disease ranging from benign unifocal bone disease to an aggressive multisystem disease. Eosinophilic granuloma, the most localised and benign form of LCH, usually presents in children less than four-years-of-age as unifocal bone disease and possible orbital involvement.5
Clinical Presentation
LCH commonly involves the superotemporal orbit, manifesting as non-axial proptosis, ptosis, erythema, and enlarging palpebral fissures. These patients must be followed because they can develop perineural and intracranial extension, which can progress to multifocal disease.5
Neuroimaging
CT demonstrates osseous destruction, whereas MRI better evaluates intracranial extension.

Figure 7. A: Postcontrast T1-weighted fat-saturated axial image shows a well-defined, lobulated intraconal mass with patchy, irregular enhancement and B: on the T2-weighted fat-saturated image the mass is hyperintense with linear low-intensity areas, likely flow voids, consistent with orbital hemangioma.17
CT
Well-defined or diffusely homogeneous soft-tissue lesions,
Replaces/destroys osseous structures, and
Moderate to marked enhancement after contrast administration.
MRI
Heterogeneous signal on T1W1,
Hyperintense or hypointense on T2WI, and
Enhancement on T1-weighted postcontrast images.
Lymphoproliferative Disease and Orbital Inflammatory Syndrome
Lymphoproliferative disease present in the first decade of life, is rare in children and represents less than 5–10% of orbital lesions. Lymphoproliferative disease can be categorised into: (1) reactive lymphoid hyperplasia, (2) atypical lymphoid hyperplasia (Figure 4) and (3) ocular adnexal lymphoma. Orbital inflammatory syndrome (OIS, Figure 5), however, tends to manifest later in teenage years.
Clinical Presentation

Figure 8. A: MR coronal T2WI and B: postcontrast axial T1-weighted fat-saturated image showing a lobulated, trans-spatial mass with fluid-fluid levels, and trace rim enhancement (white arrows), consistent with orbital lymphangioma.17
Orbital lymphoproliferative disease often presents as a supero-temporal orbital mass with dystopia due to a predilection for the lacrimal gland. Diffuse extraocular muscle involvement can mimic thyroid ophthalmopathy. Signs include gradual, painless progressive nonaxial proptosis. Up to 16% of patients with orbital lymphoproliferative disease will develop systemic lymphoma. Hence, systemic evaluation for lymphoma is an important consideration in these patients.6
In contrast, OIS is often painful, but similarly involves the lacrimal gland and/or extraocular muscles, causing non-axial proptosis.
Neuroimaging
MRI
Lymphoid hyperplasia:
Intermediate signal intensity on T1/T2W1, Moderate enhancement with gadolinium contrast.
Lymphoma:
Isointense to hypointense on T1W1, Isointense to hyperintense on T2W1.

Figure 9. A: MR postcontrast axial T1-weighted images with fat saturation demonstrating the enhancing soft-tissue mass and its extension into the medial orbit, displacing the right medial rectus laterally (black arrow), causing proptosis (star) and B: CT coronal with bone windows, demonstrating osseous destruction and invasion of the right medial orbital wall, bilateral ethmoid sinuses, right frontal sinus, both nasal cavities, turbinates, and nasal septum, consistent with a rhabdomyosarcoma.17
Orbital inflammatory syndrome:
Isointense to hypointense on T1W1, Homogenous enhancement on T2W1 with fat supression.
MRI DWI is more sensitive in discriminating between orbital lymphoproliferative disease (significantly restricted diffusion) and OIS.
VASCULAR LESIONS
Infantile Haemangioma Infantile haemangiomas (Figure 7) are hamartomas of capillary endothelial cells. They are the most common vascular tumour of childhood in the orbit and are classified into preseptal, intra-orbital (involving the post-septal orbit), and compound/mixed, involving both.7
Clinical Presentation
Most cases are diagnosed within the first weeks to months of life, after which the haemangioma begins a proliferative phase for up to 10 months. During this phase, the lesion may be complicated by haemorrhage and ulceration, causing obstruction of the visual axis, occlusion amblyopia, astigmatism, corneal ulceration, secondary glaucoma through compression of the aqueous outflow channels, and compressive optic neuropathy. Following the first year of life, growth stabilises, followed by a prolonged course involution for up to 10 years.7 Haemangiomas vary widely from superficial, flat lesions with a characteristic strawberry colour, to deeper, bluish lesions and large disfiguring masses. Intra-orbital haemangiomas can present with non-axial proptosis and demonstrate intracranial extension. Multifocal haemangiomas, seen in up to a third of patients, demonstrate involvement of the viscera and skin.8

Figure 10. A: MR axial enhanced T1 and B: coronal T2 demonstrating an ill-defined left intra-orbital mass, demonstrating enhancement and invasion of the intracranial space through the superior orbital fissure, consistent with orbital neuroblastoma.18
Neuroimaging
Ultrasound
Well-circumscribed mass and variable echogenicity,
High-flow system with low resistance, devoid of arteriovenous shunting.
MRI
Provides more sensitive evaluation of deeper lesions and adjacent structures.
MRA
Assesses flow characteristics,
Well-circumscribed solid masses with arterial flow voids,
Isointense/heterogenous signal on T1WI,
Hyperintense on T2WI,
Marked enhancement on post-contrast images, and
High flow on time-resolved MRA, with decreasing flow voids and enhancement during involution.

Figure 11. A: MR axial T2W1 demonstrating fusiform enlargement of bilateral optic nerves, right greater than left (white arrows), with associated right globe deviation, and B: postcontrast axial T1WI with fat saturation demonstrating diffuse enhancement of the optic nerves (double white arrow) with extension into the optic chiasm, consistent with optic pathway glioma.17
Orbital Lymphangioma/ Venous-Lymphatic Malformations
Orbital lymphangiomas (Figure 8) represent a spectrum of complex congenital lesions of the orbit, comprising of a mixture of maldeveloped lymphatic and vascular structures during embryologic life. They vary from superficial, largely lymphatic lesions to deep venous lesions. They represent 4% of all paediatric orbital masses and are usually evident by age two.9,10 Gradual enlargement is uncommon, whereas puberty usually results in accelerated growth.
Clinical Presentation
Superficial lesions are noted earlier and can extend to the forehead and cheek. Deeper lesions however, present later with acute non-axial proptosis secondary to haemorrhage, restricted ocular motility or acute lesion enlargement with a concomitant upper respiratory tract infection. Haemorrhage results in variable-sized chocolate-coloured cysts occurring.9
Periorbital lesions have a more severe presentation including blepharoptosis, fluctuating non-axial proptosis, intermittent lid swelling, intralesional bleeding, pain, amblyopia, chemosis, astigmatism, restrictive strabismus, exposure keratopathy and compressive optic neuropathy. As a result, up to 40% of children can have vision reduction in the affected eye.9
Orbital lymphangiomas are associated with intracranial vascular anomalies in up to 70% of patients, and imaging of the brain should be performed concurrently.9
Neuroimaging

Figure 12. MR axial T1 weighted image demonstrating a homogenous isointense mass in the right orbit, indicative of a granulocytic sarcoma.19
MRI
Irregular, lobulated, infiltrating with ill-defined margins (due to lack of a capsule),
Involves the pre/post-septal and intra/extraconal portions of the orbit,
Solid appearing microcysts, macrocysts can measure up to 2cm,
Fluid levels, due to intralesional haemorrhage, are almost pathognomonic,
Lack of flow voids (differentiating feature from high flow lesions),
Isointense signal on T1WI,
Hyperintense with presence of internal septations on T2WI, and
Heterogenous enhancement on post-contrast images (venous component enhances and lymphatic component shows fine enhancement of septations).
MALIGNANT LESIONS
Rhabdomyosarcoma
Orbital rhabdomyosarcoma (Figure 9) is the most common paediatric orbital malignancy, presenting at a mean age of six to eight-years-of- age. Rhabdomyosarcoma is a soft-tissue sarcoma with two major subtypes in the orbit: 1) alveolar (less common, high risk), and 2) embryonal (more common, intermediate risk).10 There is a known association in children with the following inherited diseases: Li-Fraumeni Syndrome, Beckwith- Widemann Syndrome, Neurofibromatosis type 2, Noonan Syndrome, and Multiple Endocrine Neoplasia type 2a Syndrome.
Clinical Presentation
Orbital rhabdomyosarcoma presents as a rapidly growing, painless mass, most commonly supero-medial (embryonal) versus inferior (alveolar), resulting in non-axial proptosis. Anterior tumour involvement into the levator palpebrae superioris can result in eyelid oedema, haemorrhage, pain, isolated blepharoptosis, and chemosis. Posterior involvement can result in a compressive optic neuropathy and subsequently, compromise vision.10 The rapid growth and aggressive nature frequently results in invasion of adjacent bone and soft tissue structures. Fortunately, local lymph node metastases and intracranial invasion are relatively uncommon. Distant metastases most often involve the lungs and bones. A systemic evaluation is therefore crucial.10
Neuroimaging
MRI
Isointense to muscle on T1WI,
Hyperintense to muscle on T2WI,
Moderate to marked enhancement postcontrast on T1W1 with fat saturation, and
Globe and extra-ocular muscle displacement may be noted but invasion is rare, and intracranial or adjacent paranasal sinus invasion is best seen on comparison of pre- and post-contrast T1WI images.
CT
Isoattenuation to muscle,
Moderate to marked enhancing extraconal, homogeneous, and well-circumscribed mass,
Calcifications occurring with bony destruction, and
Best demonstrates bony destruction and complements the MRI soft tissue findings in cases of diagnostic uncertainty.11
Neuroblastoma
Neuroblastoma (Figure 10) is the most frequent extracranial solid tumour of childhood, arising from neural crest cells. It is the most common primary childhood cancer to metastasise to the orbits. Of all cases, 20% have orbital involvement, which frequently is the primary manifestation. Orbital neuroblastoma is a disease of the very young, with a reported median age of two years.12
Clinical Presentation
Orbital neuroblastoma presents classically as unilateral/bilateral non-axial proptosis and periorbital/eyelid ecchymosis (raccoon eyes). Less frequent clinical findings include periorbital swelling, haemorrhage, ptosis, restrictive strabismus and optic nerve atrophy.12-13 Opsoclonus, characterised by rapid, multidirectional saccadic eye movements, is a paraneoplastic syndrome associated with orbital and systemic neuroblastoma. This is generally a good prognostic factor, but neurologic deficits may remain. Horners syndrome (ptosis/inverse ptosis, miosis and infrequently anhidrosis) only occurs in the context of extra-orbital neuroblastoma due to its predilection for the sympathetic chain.
Neuroimaging
MRI
Hypointense on T1WI,
Heterogeneous on T2WI due to haemorrhage or necrosis, and
Heterogeneous enhancement postcontrast.
Optic Pathway Glioma
Optic pathway gliomas (Figure 11) can involve any portion of the visual pathway, including the hypothalamus, optic disc, nerve, chiasm, geniculate nucleus, and optic radiations. They most frequently present in the first decade, with a mean age between six and seven-years-of-age. The incidence is highest in children with neurofibromatosis 1 (NF1), an autosomal dominant mutation, ranging from 30-58%. Symptomatic lesions, however, only occur in 1–5% of patients. Bilateral involvement of the optic pathways is pathognomonic for NF1. Histology typically shows low-grade gliomas (WHO grade I-II), but disease progression can be varied.14-15
Clinical Presentation
Optic pathway gliomas are slow growing lesions and therefore can go unrecognised clinically, presenting with slowly progressive vision loss. Clinical signs include reduced visual acuity, colour vision, visual field defects, relative afferent pupillary defect (RAPD), axial proptosis, ocular motility dysfunction and optic disc oedema/pallor/atrophy. Hypothalamic lesions can cause hydrocephalus, where 15-year mortality rates approach 50%.14-15
Neuroimaging
MRI
Findings are characteristic and specific, more sensitive for detection of smaller lesions and intracranial extension,16
Fusiform enlargement of the optic nerve,
Widening of the optic canal, Variable contrast enhancement, Isointense or hypointense signal on T1WI, Hyperintense signal on T2WI,
Variable enhancement on postcontrast images,
Increased signal on post-contrast images with T2WI/FLAIR hyperintensity indicating posterior extension, and
Differentiation from optic nerve meningiomas: hypointense on T2WI and avidly enhanced on post-contrast images.
Granulocytic Sarcomas
Granulocytic sarcomas (Figure 12), a rare solid tumour made of primitive granulocyte precursors within the orbit, are most prevalent among paediatric patients with acute myeloid leukaemia (AML). Mean age of presentation is between age seven and nine years, with 90% of cases reported to have unilateral involvement.17 It can also occur in association with chronic myeloid leukaemia (CML), myelofibrosis with myeloid metaplasia, hypereosinophilic syndrome and polycythaemia vera.
Clinical Presentation
These lesions are largely asymptomatic. In a patient with known leukaemia, suspicious clinical features include a rapidly enlarging orbital mass and non-axial proptosis, pain, eyelid swelling, ecchymosis and diplopia, mimicking orbital cellulitis. Presentation can be prior to, or following the diagnosis of AML, and can indicate relapse in treated patients.17 It is associated with a poor prognosis when noted in children without a background of haematological disease.
Neuroimaging
MRI
Irregular homogenous masses,
Lateral orbital predilection, encasing the adjacent lacrimal gland and extraocular muscles,
Isointense or hypointense to muscle on T1W1,
Heterogeneously isointense or hyperintense, on T2W1, and
Homogenous enhancement on postcontrast sequences.
CONCLUSION
Paediatric orbital lesions present a diagnostic challenge. Given the overlapping and complex spectrum of clinical presentation, characteristic neuro-imaging features are crucial to facilitate an accurate diagnosis. A systematic, targeted multidisciplinary approach is key in the initiation of timely treatment to minimise the potential morbidity and mortality associated with these lesions in children.
To earn your CPD hours from this article visit mieducation.com/paediatric-ocular-tumoursclinical- presentations-and-neuroimaging.
 Dr Sonia Moorthy MBChB (University of Dundee), MPH (University of Melbourne), FRANZCO is an adult and paediatric ophthalmologist. Her ophthalmic training was completed at the Sydney Eye Hospital. She subsequently completed fellowships, in paediatric ophthalmology and strabismus, at the prestigious Singapore National Eye Centre and Moorfields Eye Hospital, London.
Dr Sonia Moorthy MBChB (University of Dundee), MPH (University of Melbourne), FRANZCO is an adult and paediatric ophthalmologist. Her ophthalmic training was completed at the Sydney Eye Hospital. She subsequently completed fellowships, in paediatric ophthalmology and strabismus, at the prestigious Singapore National Eye Centre and Moorfields Eye Hospital, London.
Dr Moorthy has recently moved to the Sunny Coast from Far North Queensland. She continues to provide Indigenous eye care as a visiting ophthalmologist to Emerald, Central Queensland. She is also committed to training the next generation of ophthalmologists and sits within the RANZCO Ophthalmic Sciences Board of Examiners and Women in Ophthalmology committee group.
References
- E. M. Chung, M. D. Murphey, C. S. Specht, R. Cube, and J. G. Smirniotopoulos, From the archives of the AFIP pediatric orbit tumors and tumorlike lesions: osseous lesions of the orbit. Radiographics, vol. 28, no. 4, pp. 1193–1214, 2008.
- T. S. Wells and G. J. Harris, Orbital dermoid cyst and sinus tract presenting with acute infection. Ophthalmic Plastic and Reconstructive Surgery, vol. 20, no. 6, pp. 465–467, 2004.
- C. Jacquemin, T. M. Bosley, and H. Svedberg, Orbit deformities in craniofacial neurofibromatosis type 1. American Journal of Neuroradiology, vol. 24, no. 8, pp. 1678–1682, 2003.
- J. J. Dutton and G. K. Escaravage, Ophthalmic Oncology, edited by B. Esmaeli, Springer, Boston, Mass, USA, 2011.
- H. Vosoghi, C. Rodriguez-Galindo, and M. W. Wilson, Orbital involvement in langerhans cell histiocytosis. Ophthalmic Plastic and Reconstructive Surgery, vol. 25, no. 6, pp. 430–433, 2009.
- H. Demirci, C. L. Shields, E. C. Karatza, and J. A. Shields, Orbital lymphoproliferative tumors: analysis of clinical features and systemic involvement in 160 cases. Ophthalmology, vol. 115, no. 9, pp. 1626–1631, 2008.
- B. G. Haik, F. A. Jakobiec, R. M. Ellsworth, and I. S. Jones, Capillary hemangioma of the lids and orbit: an analysis of the clinical features and therapeutic results in 101 cases. Ophthalmology, vol. 86, no. 5, pp. 760–789, 1979.
- W. R. K. Smoker, L. R. Gentry, N. K. Yee, D. L. Reede, and J. A. Nerad, Vascular lesions of the orbit: more than meets the eye. Radiographics, vol. 28, no. 1, pp. 185–204, 2008.
- A. Bisdorff, J. B. Mulliken, J. Carrico, R. L. Robertson, and P. E. Burrows, Intracranial vascular anomalies in patients with periorbital lymphatic and lymphaticovenous malformations. American Journal of Neuroradiology, vol. 28, no. 2, pp. 335–341, 2007.
- J. A. Shields, C. L. Shields, and R. Scartozzi, Survey of 1264 patients with orbital tumors and simulating lesions: the 2002 Montgomery Lecture.
- N. J. M. Freling, J. H. M. Merks, P. Saeed et al., Imaging findings in craniofacial childhood rhabdomyosarcoma. Pediatric Radiology, vol. 40, no. 11, pp. 1723–1738, 2010.
- J. C. Gutierrez, A. C. Fischer, J. E. Sola, E. A. Perez, and L. G. Koniaris, Markedly improving survival of neuroblastoma: a 30- year analysis of 1,646 patients. Pediatric Surgery International, vol. 23, no. 7, pp. 637– 646, 2007.
- N. D’Ambrosio, J. Lyo, R. Young, S. Haque, and S. Karimi, Common and unusual craniofacial manifestations of metastatic neuroblastoma. Neuroradiology, vol. 52, no. 6, pp. 549–553, 2010.
- M. V. Mishra, D. W. Andrews, J. Glass et al., Characterization and outcomes of optic nerve gliomas: a population-based analysis. Journal of Neuro-Oncology, vol. 107, no. 3, pp. 591–597, 2012.
- A. G. Lee and J. J. Dutton, A practice pathway for the management of gliomas of the anterior visual pathway: an update and an evidence-based approach. Neuro- Ophthalmology, vol. 22, no. 3, pp. 139–155, 1999.
- S. C. Jost, J. W. Ackerman, J. R. Garbow, L. P. Manwaring, D. H. Gutmann, and R. C. McKinstry, Diffusionweighted and dynamic contrast-enhanced imaging as markers of clinical behavior in children with optic pathway glioma. Paediatric Radiology, vol. 38, no. 12, pp. 1293–1299, 2008.
- A. A Rao, J. H. Naheedy et al., A clinical update and radiologic review of pediatric orbital and ocular tumors. J Oncol. 2013; doi: 10.1155/2013/975908.
- S. T. Hlongwane, M. Pienaar, G. Dekker, A. Brandt, D. van der Merwe, H. B. Louw, S. Dajee. Proptosis as a manifestation of neuroblastoma. SA Journal Of Radiology December 2006.
- Stockl FA, Dolmetsch AM, Saornil MA, Font RL, Burnier MN Jr. Orbital granulocytic sarcoma. Br J Ophthalmol. 1997 Dec;81(12):1084-8. doi: 10.1136/bjo.81.12.1084. PMID: 9497470; PMCID: PMC1722070.