
Unusual corneal dots, lines, and shadows on the cornea can be intimidating to a practitioner when trying to identify whether a patient is experiencing a corneal dystrophy or degeneration.
CORNEAL DYSTROPHIES
Corneal dystrophies are characterised by the layer that they are found in, and will show as specific bilateral, usually symmetrical opacities. Initially presenting in younger people in the central cornea, they become denser and extend to the corneal periphery over time. Fortunately, some corneal dystrophies remain small and do not affect vision early on. Most slowly progress, and as these opacities enlarge, they become more visually debilitating. Early corneal dystrophies can benefit from specialty contact lenses to minimise disturbance to vision in early stages. Surgical intervention is often required at later stages of the disease.
Due to an autosomal dominant inheritance, all family members of patients with corneal dystrophies should be referred for testing and early diagnosis
 Being autosomal dominant, family members of those with corneal dystrophy should be encouraged to be tested – as many as 50% will have the same condition.1
Being autosomal dominant, family members of those with corneal dystrophy should be encouraged to be tested – as many as 50% will have the same condition.1
Corneal dystrophies are rarely associated with systemic disease and are noninflammatory. The presence of corneal neovascularisation indicates the condition is not a corneal dystrophy, but rather, another pathophysiological process, potentially including a corneal degeneration.
CORNEAL DEGENERATIONS
Corneal degenerations differ from corneal dystrophies in that they tend to be asymmetric opacities in the periphery, and result from ageing, metabolic or inflammatory changes.
Corneal degenerations include:
- Crocodile shagreen – a greyish, polygonal pattern of opacities with intervening clear zones across the central cornea that resemble crocodile skin.
- Vogt’s girdle – a thin, whitish-yellow band at the temporal limbus, with a clear rim of cornea between band and limbus.
- Arcus senilis.
- Band keratopathy – inflammatory fine dust-like calcium deposits in the sub-epithelium, Bowman’s layer and the anterior stroma. It is typically a band-shaped, horizontal opacity that grows from the peripheral cornea towards the central cornea.
- Salzmann’s nodular degeneration – slowly progressive elevated nodules near the limbus to mid peripheral cornea, seen anterior to Bowman’s layer of the cornea.
CORNEAL DYSTROPHY IDENTIFICATION AND CATEGORISATION
As discussed, corneal dystrophies are characterised by the layer they are found in. Superficial corneal dystrophies are those considered to affect the corneal epithelium, Bowman’s layer and anterior stroma. These are responsive to treatment with less invasive surgical procedures than a full penetrating keratoplasty or deep lamellar keratoplasty. Early identification is helpful for improved prognosis and prevention of vision loss. Phenotypical identification can be helpful in this categorisation process.
Good biomicroscopic and illumination techniques, and anterior optical coherence tomography (OCT), can help in the identification process. Illumination techniques include:
- Direct illumination to identify the number, types and location of the corneal opacification.
- Creation of an optic section/parallelepiped to identify the corneal layer affected.
- Indirect illumination – retroilluminate and sclerotic scatter to identify small early lesions missed on first glance.
EPITHELIAL DYSTROPHIES
Corneal dystrophies affecting the corneal epithelial layer include:
- Epithelial basement membrane dystropy (EBMD) – previously called map-dot fingerprint dystrophy – is characterised by lesions appearing as small continents on a map, fingerprint swirls, small dot or milky white bleb opacities.
- Meesmann dystrophy – characterised by early presentation, diffuse, tiny greywhite vesicles, extending to the limbus as the vesicles, burst, and causing recurrent corneal erosions.
- Lisch corneal dystrophy – previously called whirled, band-shaped, microcystic dystrophy – is classified by patterns of feathery-shaped linear whirled opacities, small depositions in the epithelial cells. This dystrophy is considerably helped by rigid gas permeable lenses.
- Epithelial recurrent corneal erosion dystrophy (ERED) – presenting as young as age five, and characterised by aggressive recurrent corneal erosions with multiple triggers of smoke, dry air, upper respiratory tract infections and minimal trauma. Repeated erosions unfortunately cause corneal keloid scarring, which require corneal transplantation in approximately 25% of cases.2
Recurrent corneal erosions linked with epithelial dystrophies can be treated with antibiotics, artificial tears and gels, bandage contact lenses, phototherapeutic keratectomy, debridement, stromal puncture, and amniotic membrane therapy.
BOWMAN’S LAYER DYSTROPHIES
Bowman’s layer dystrophies include:
- Reis-Buckler corneal dystrophy (RBCD) – the most common Bowman’s layer dystrophy – presenting with ring-shaped opacities with localised areas of Bowman’s layer thickening, resulting in epithelial irregularity, irregularity astigmatism and recurrent corneal erosions. Contact lenses, superficial keratectomy, penetrating keratoplasty and mitomycin C can help prevent recurrences.
- Thiel-Benkhe dystrophy (TBCD) affects Bowman’s layer, similar to RBCD though less progressive, and may be an RBCD variant.
ANTERIOR STROMAL DYSTROPHIES
Stromal dystrophies can also cause epithelial and structural changes, and epithelial erosions. The more anterior stromal dystrophies are amenable to phototherapeutic keratectomy.
Stromal dystrophies include:
- Granular dystrophy, characterised by granular central opacities made of hyaline, slowly progressing to cause slow deterioration of vision.3
- Lattice dystrophy, characterised by a latticelike network of refractile lines in the anterior stroma, composed of amyloid plaques, spreading from the central cornea to the corneal periphery.
- Combined granular-lattice dystrophy (Avellino corneal dystrophy), reported first in the town of Avellino, Italy, with granular opacities in younger, and lattice lines in older, individuals.4
- Crystalline dystrophy of Schnyder – a stromal dystrophy – characterised by crystalline, refractile deposits in the stroma, composed of cholesterols and phospholipids, with arcus presenting concurrently, and a low impact on vision. Check for systemic dyslipidemia if seen in younger patients.5
In early cases, contact lenses and, for more advanced cases, phototherapeutic keratectomy can be useful in the treatment of these dystrophies.
POSTERIOR CORNEAL DYSTROPHIES
Posterior corneal dystrophies include:
- Fuch’s endothelial dystrophy – affecting women slightly more than men – generally has a later onset but there is also an early-onset hereditary variant (20–40 year-olds). Early-onset Fuchs dystrophy is characterised by corneal guttata in Descemet’s membrane causing endothelial compromise, epithelial and stromal oedema, and eventual corneal fibrosis. Treatment is with hyperosmotic agents and nightly ointment. Surgical intervention for more severe cases is indicated with deep lamellar endothelial keratoplasty, Descemets stripping endothelial keratoplasty, and penetrating keratoplasty.
- Posterior polymorphous dystrophy (PPD) presents with vesicle-like lesions, bands or diffuse opacities appearing as thickening of Descemet’s membrane. PPD can result in adhesions between the iris and the cornea (peripheral anterior synechiae), and is consequently a risk for glaucoma.6
- Congenital hereditary endothelial dystrophy (CHED) – an endothelial dystrophy – causes corneal oedema from infancy or birth. It also presents with nystagmus, and is treated with penetrating keratoplasty.
CASE STUDY
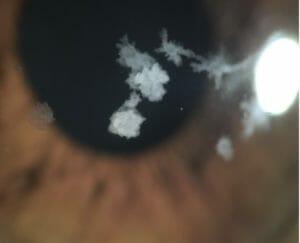
Figure 1. Xavier’s corneal dystrophy.
Xavier,* a 37-year-old male construction engineer, was referred by his corneal specialist for specialty contact lens fitting. Xavier was referred due to the corneal irregularities caused by his corneal dystrophy. Symptoms caused by the dystrophy included glare, blurry vision, headaches and eyestrain associated with computer work.
Slitlamp biomicroscopy showed bilateral granular stromal lesions characteristic of granular stromal dystrophy (Figure 1).
Entering VA 6/15, 6/18, OU 6/9.
He was fitted with semi-scleral RoseK2 XL contact lenses, which yielded vision of RE 6/7.5; L 6/9 OU 6/7.5.
Spectacles yielded vision of RE 6/12 LE 6/12 OU 6/7.5.
With his specialty contact lenses, Xavier noticed less glare and headaches. Overall, he reported his vision to be considerably better.
Xavier now alternates between glasses and contact lens correction, depending on preference and convenience. He is able to obtain satisfactory vision with his spectacle and contact lens options for now, without more invasive treatments. His sibling and father also had the exact same condition, consistent with a high familial link with corneal dystrophies.
TREATMENT CONSIDERATIONS FOR CORNEAL DYSTROPHIES
Specialty contact lenses, antibiotics, artificial tears and gels, bandage contact lenses, phototherapeutic keratectomy, corneal debridement, stromal puncture, and amniotic membrane therapy can smooth the irregular corneal surface and remove or reduce the density of the corneal opacities in corneal dystrophies. These treatments, depending on the severity of the patient’s presenting condition, should be considered for patients who present with corneal dystrophies. Due to an autosomal dominant inheritance, all family members of patients with corneal dystrophies should be referred for testing and early diagnosis – this will improve prognosis for patients and prevent potential sight loss.
*Patient name changed for anonymity. Dr Margaret Lam is the National President of Optometry Australia and a Director of Optometry NSW/ACT. She teaches at the School of Optometry at UNSW as an Adjunct Senior Lecturer and works as the Head of Optometry Services for George and Matilda Eyecare.
References
- www.reviewofoptometry.com/article/the-genetics-ofcorneal- dystrophies#:~:text=Most%20dystrophies%20 are%20inherited%20as,some%20dystrophies%20are%20 sex%2Dlinked.
- Hammer B, Bjorck E, Langenstedt K, et al. A new corneal disease with recurrent erosive episodes and autosomal dominant inheritance. Acta Ophthalmol. 2008;86(7):758-63.
- Groenouw A. Knotcchenformige Hornhauttrubungen. Arch Augenheld. 1890;21:281-89.
- Kennedy SM, McNamara M, Hillery M, et al. Combined granular lattice dystrophy (Avellino corneal dystrophy). Br J Ophthalmol. 1996;80(5):489-90.
- Garner A, Tripathi PC. Hereditary crystalline corneal dystrophy of Schnyder II. Histopathology and ultrastructure. Br J Ophthalmol 1972;56(5):400-8.
- www.reviewofoptometry.com/article/the-genetics-ofcorneal- dystrophies#:~:text=Most%20dystrophies%20 are%20inherited%20as,some%20dystrophies%20are%20 sex%2Dlinked.





