Monitoring a patient with granular corneal dystrophy has given Jessica Chi cause to review corneal dystrophies – rare, inherited conditions that affect corneal transparency.
Diego,* a 36-year-old male, who had emigrated to Australia eight years prior, presented reporting no noted problems with his vision. His sister in Spain had some visual issues and she had been advised that family members should be tested for the same condition. He denied any ocular problems and had never had his eyes tested.
Vision unaided was R and L 6/6, correcting to R and L 6/4.8= with a mild astigmatic correction.
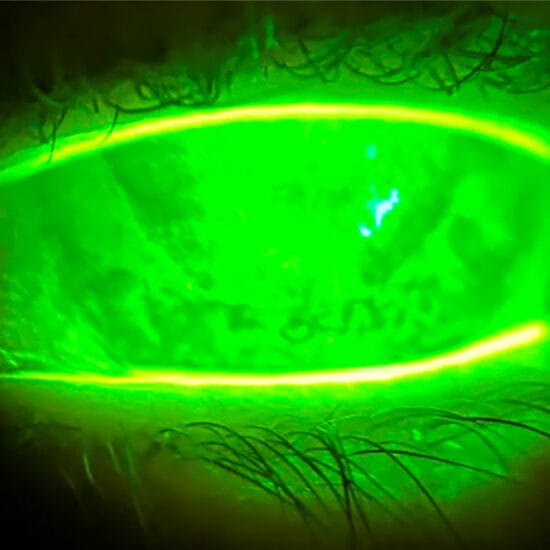
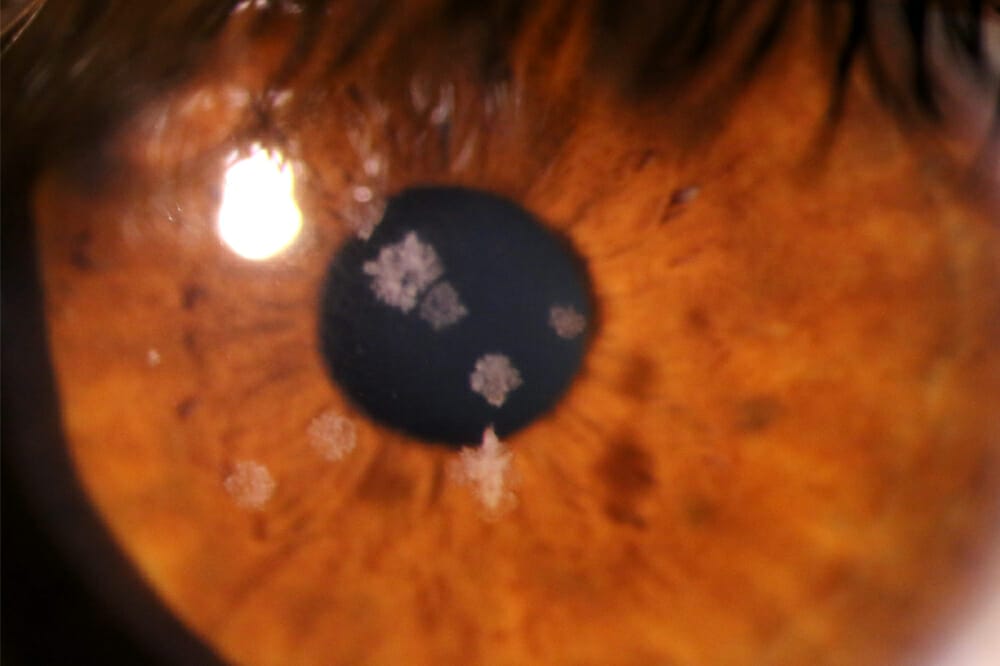
Anterior eye exam revealed discrete, multiple, fluffy but well demarcated corneal stromal lesions in both eyes (Figure 1), concentrated centrally. The corneal epithelium was intact, and there was no staining or active inflammation noted.
Diego was diagnosed as having granular corneal dystrophy. An initial review was set for 12 months. Since then, Diego has been reviewed for a further four years. Most recently, he reported that his sister’s condition has deteriorated and she may have a corneal transplant. Fortunately, Diego’s visual acuity has only dropped one or two letters on the Snellen chart since we began monitoring him.
DISCUSSION
Corneal dystrophies are rare, inherited conditions that affect corneal transparency due to the accumulation of abnormal material in the cornea. Bilateral, non-inflammatory, and progressive; they result in opacities in the cornea of various conformations, primarily affecting the central cornea. Changes often begin in one of the layers in the cornea and can spread to other layers.
Corneal dystrophies are differentiated from degenerations in that they are inherited, bilateral, symmetric, insidious in presentation, and unrelated to environmental factors. Most corneal dystrophies are inherited in an autosomal dominant pattern, but some are autosomal recessive or X-linked, and some studies suggest they may be the result of consanguinity.
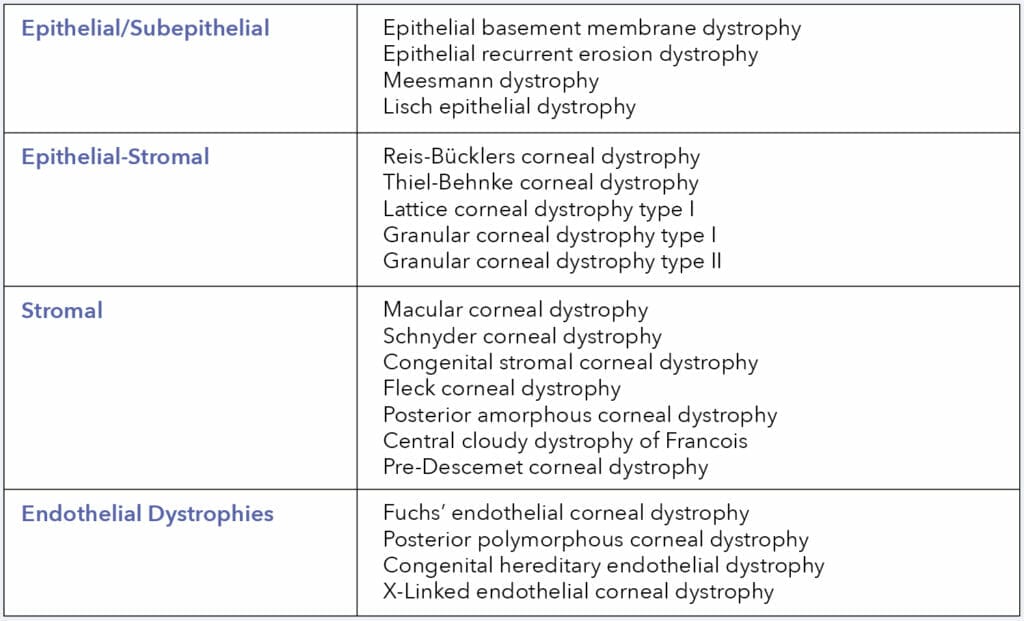
Corneal dystrophies may be sub-classified by the anatomic location affected – as proposed by the International Classification of Corneal Dystrophies – epithelial/subepithelial, epithelial-stromal, stromal, and endothelial dystrophies.
In the early stages, patients may be asymptomatic. However, as the condition progresses, they may suffer from visual symptoms such as bilateral reduction in visual acuity and photophobia.
Corneal dystrophies are rare, inherited conditions that affect corneal transparency due to the accumulation of abnormal material in the cornea
Signs and symptoms are related to the anatomical location of the dystrophy:
- Epithelial and subepithelial dystrophies commonly present with corneal erosions, especially upon waking. Management can include artificial tears, hypertonic nighttime lubricants, and bandage contact lenses. For more severe erosions, superficial keratectomy or photokeratectomy (PTK) may be indicated.
- Stromal dystrophies commonly present with dry eye, corneal oedema, and corneal erosions, particularly in those with epithelial disease.
The optometric management of many of these dystrophies can present a challenge to the clinician. Treatment may range from simple therapeutic contact lenses to referral for penetrating keratoplasty.

Anterior eye exam of a left eye (A) and right eye (B)
Granular corneal dystrophy is autosomal dominant, with the genetic locus on 5q31, and the onset in childhood. In childhood, it may present as a vortex pattern of brownish granules developing superficial to Bowman’s layer. With time, demarcated white granules develop with clear intervening stroma. These can progress, resulting in a snowflake appearance. On retro-illumination, they are composed of small, translucent dots, which appear like crushed breadcrumbs or glassy splinters. The opacities tend to be central and respect the limbus. Later, granules may extend to the deeper stroma approaching Descemet’s membrane.
In the early stages, patients may present with glare and photophobia. As the condition progresses, acuity reduces.
STROMAL DYSTROPHIES
Lattice Corneal Dystrophy
Type 1 is characterised by the presence of thin, branching refractile lines within the stroma, sparing the limbus and gradually leading to generalised stromal haze. Histology shows amyloid deposits in the stroma, which stain with Congo red. The lines start appearing at the end of the first decade; centrally and superficially at first, then spreading centrifugally. Type 1 then involves the deeper layers, sparing the peripheral 1mm, Descemet’s membrane and endothelium. It is inherited in an autosomal dominant pattern; the defect is linked to the TGFB1 gene.
Type 2, or Meretoja syndrome, is characterised by sparse lines. There are features of systemic amyloidosis with bilateral cranial and peripheral neuropathy. The corneal lines spread centripetally from the limbus, and the central cornea is spared.
Type 3 is characterised by late onset, at the age of 70–90 years, with thick rope-like bands.
Granular Dystrophy
This begins in childhood and is characterised by small, white, sharply demarcated deposits resembling crumbs. Initially, the stroma between the opacities is clear, but the deposits gradually increase in size, number, and confluence, leading to diffuse haze. The limbus is typically spared. Histology shows hyaline deposits staining with Masson’s trichrome. This is another corneal dystrophy linked to the TGFB1 gene. It is inherited in an autosomal dominant pattern.
Avellino Dystrophy
This is a combined granular and lattice dystrophy. Histology shows both hyaline and amyloid deposits in the stroma. It usually manifests in the second decade, with refractile lines and granular deposits. Recurrent erosions are rare.
Macular Dystrophy
This dystrophy represents an in-born error of keratin sulphate metabolism. Onset is at
the end of the first decade and patients usually present with reduced vision. It is inherited in an autosomal recessive pattern. The genetic defect is on the carbohydrate sulfotransferase 6 (CHST6) gene. Clinically there are poorly delineated deposits in the central corneal stroma with surrounding anterior stromal haze. The deposits gradually increase in size, involving the full thickness of stroma up to the limbus. Histology shows deposits of glycosaminoglycan in the stroma, which stains with alcian blue.
Schnyder Crystalline Corneal Dystrophy
This dystrophy represents a defect in corneal lipid metabolism. Onset is in the second decade, and 50% of patients have raised serum cholesterol levels. Histology shows deposits of phospholipids and cholesterol. It is characterised by subepithelial crystalline deposits. Eventually, by the third decade there is diffuse corneal haze and prominent corneal arcus. The genetic defect is on the UB1AD1 gene with autosomal dominant inheritance.
Central Cloudy Dystrophy of Francois
This has similar appearance to crocodile shagreen with polygonal cloudy grey opacities in the posterior stroma giving a ‘leather-like appearance’. The difference, however, is that unlike crocodile shagreen, Francois dystrophy is in a central and posterior location. It is inherited in an autosomal dominant pattern. Patients are usually asymptomatic.
Congenital Stromal Corneal Dystrophy
Otherwise known as Whitschel dystrophy, this dystrophy is very rare. Present at birth, it consists of numerous opaque, flaky, or feathery areas of clouding in the stroma that multiply with age. There is some association with primary open glaucoma. The mutation is on the DCN gene on chromosome 12q22, which encodes the protein decorin. It is inherited in an autosomal dominant manner.
Jessica Chi is the director of Eyetech Optometrists, an independent specialty contact lens practice in Melbourne. She is the current Victorian, and a past national president of the Cornea and Contact Lens Society, and an invited speaker at meetings throughout Australia and beyond. She is a clinical supervisor at the University of Melbourne, a member of Optometry Victoria Optometric Sector Advisory Group and a Fellow of the Australian College of Optometry, the British Contact Lens Association and the International Academy of Orthokeratology and Myopia Control.
Reference
Weiss J.S., Møller H.U. et al. IC3D classification of corneal dystrophies – edition 2. Cornea. 2015 Feb;34(2):117-59.