The ability to accurately diagnose genetic ophthalmic conditions is critical to facilitating effective counselling, disease treatment and management.
The genomic medicine revolution is providing answers for many families with inherited eye disease. The implementation of next generation genetic screening starting to be introduced into clinical practice is the principal enabler for these advances. Where previously only one gene could be tested at a time, now many hundreds can be tested simultaneously. This ability is extremely helpful in inherited eye diseases since both the retinal disorders and the developmental eye, including congenital cataract, are very heterogenous conditions. This means that many different genes can produce the same clinical features, making selecting a single gene to test almost impossible.
The power of the technology is also adding to the complexity when interpreting the genetic results. Genetic variants are being identified in individuals in both known and novel genes. The clinical challenge is to determine whether these variants of uncertain significance are in fact disease causing or just another normal genetic variant (polymorphism) that may exist in the unaffected community.
Congenital Cataracts
Congenital cataracts are responsible for approximately 10 per cent of childhood blindness worldwide1 with over 50 per cent of childhood cataract cases believed to be familial and associated with 110 genes.2 Other causes including antenatal infection, trauma and chemical or drug exposure.2
Where previously only one gene could be tested at a time, now many hundreds can be tested simultaneously
In the UK, the incidence of congenital cataracts presenting in the first year of life has been estimated at 2.49 per 10,000. Prevalence rises to 3.46 per 10,000 by the age of 15 years.1 The condition can lead to life-long vision loss and is often associated with other conditions such as glaucoma, microcornea and anterior segment dysgenesis.
Understanding the underlying cause of congenital cataracts is helpful when providing information to parents and particularly when facilitating genetic counselling. It is also important for minimising unnecessary investigations and planning treatment and disease management. Yet according to M. Musleh et al, a “disappointing” number of patients with congenital cataracts received diagnosis following standard treatment.
Routine investigations for congenital cataract include TORCH screen testing for infections such as Rubella, toxoplasmosis, cytomegalovirus, herpes simplex virus one and two, syphilis and parvovirus; karyotyping and urinary and plasma organic acids. These investigations were shown to be less than satisfactory in determining the underlying cause of bilateral childhood cataracts. Indeed, the researchers questioned whether a Torch screen should be undertaken where a patient shows no other clinical indications.2 However researchers have demonstrated that next generation sequencing (NGS), is far more successful. NGS – described as a “high throughput sequencing technique that enables the parallel sequencing of multiple genes” – improved diagnostic rates, time to diagnosis and clinical management.2 In fact, NGS identified the cause of 70 – 80 per cent of single-gene disorders causing bilateral childhood cataract.2
M. Musleh et al concluded that while NGS cataract panel testing “has the power to offer considerably greater diagnostic efficacy” a realistic assessment of its value for money should be undertaken. Additionally, consideration should be given to potential negative impacts on parents receiving an early diagnosis through this screening process.
A paper by Alan S Ma et al, highlighted that a targeted NGS strategy is particularly valuable for diagnosing sporadic congenital cataract patients which make up the majority of congenital cataract cases. Ma points out that the three possible genetic mutations responsible for sporadic congenital cataract have vastly different recurrence risk likelihoods. Results from testing would therefore be useful for couples with an affected child, as well as the affected individuals who may seek accurate recurrence and transmission risk counselling.
Ma writes, “A high de novo mutation rate in this condition is important information for couples seeking reproductive information after an affected child. It indicates a low recurrence rate, with a small chance of gonadal mosaicism, and may decrease parental anxiety when planning for another child. Prenatal testing can be offered to exclude gonadal mosaicism, further enhancing reproductive confidence. In addition, probands with de novo heterozygous autosomal-dominant mutations do themselves have a 50 per cent chance of passing on their mutation to future offspring, whereas females with de novo X-linked mutations are at risk of more severely affected male offspring or recurrent miscarriages in the case of BCOR mutations. In both cases, affected individuals may wish to utilise reproductive options such as prenatal testing or preimplantation genetic diagnosis.”1
Comment
Congenital cataracts are one of the important causes of vision impairment in children. To date management has involved an attempt to identify an aetiology for the cataract but most of the clinical care has focused on surgery, optical correction and the long-term challenges of amblyopia management as well as the glaucoma risks.
The aetiology of cataracts varies. Traditionally cataracts have been thought to broadly be due to genetic aetiology; with metabolic or infectious causes together accounting for 50 per cent of cases and the remainder being idiopathic. The spectrum varies between countries with infectious causes such as rubella proportionally more common in developing countries. Rapid advances in next generation genetic sequencing have heralded in a new era in assessing the aetiology for congenital cataracts. For the first time, all the known cataract genes can be assessed simultaneously, greatly increasing the ability to identify a causative mutation. These advances are beginning to enable genotype phenotype relationships to be studied. Understanding how particular genes cause cataract as well as affect the developing eye overall will assist identifying those who will be predisposed to longer term complications including glaucoma. Additionally, the ability to test all the cataract genes, which include both syndromic and non-syndromic, can greatly assist in managing the child’s overall health. Some syndromic cataracts have only subtle systemic features or very mild dysmorphism, making identification of a systemic syndrome difficult. The genetic tests now available enhance the ability to identify early a systemic syndrome.
Beta Blocker Treatment for Infantile Hemangiomas
Infantile hemangiomas – described as benign vascular neoplasms that have a characteristic clinical course marked by early proliferation and followed by spontaneous involution3 – are common, particularly in female, white children of low birthweight.4
Hemangiomas lesions are categorised as superficial (only within the dermis), deep (involves adipose tissue), or mixed (involving multiple layers).
Until recently ophthalmic infantile hemangiomas were treated with oral corticosteroids and only when they caused functional problems by distortion of facial features, compression of the globe or closure of lids which may all be amblyogenic. Now, following the realisation that oral propranolol – a beta blocker – is a useful effective treatment with reduced side effects, treatment is being considered for cosmetic purposes or to reduce permanent scarring.4
A review of treatments for infantile hemangiomas by Sally L Painter et al, found the use of topical beta blockers was “highly successful” in stopping growth or reducing the size of the lesion.
Painter’s review found that lesions were notably softer, less purple in colour and less elevated after just a few days of treatment with oral propranolol. Lesions then began to reduce in size until they were completely flat or had disappeared. She said the natural history of the growth suggested hemangiomas treated in the proliferative stage would yield the most satisfactory results. “As the lesion is growing, there is potential to arrest development, reduce the size of the lesion, reduce its impact on surrounding structures, and ultimately improve the cosmesis of the lesion,” she wrote.4
However, she noted that if treatment was suddenly stopped, the size of the lesion may increase. Therefore, it was recommended that patients be weaned off their medication.4
The author wrote that caution should be exercised when using timolol in the periorbital area as increased absorption via the conjunctiva risked side effects.
Comment
The use of Beta blockers in managing capillary hemangiomas has been one of the great advances in the last decade. The discovery of the benefits of propranolol, a beta blocker in inducing early regression of the hemangiomas, was initially made serendipitously in infants requiring propranolol therapy for a cardiac problem who also had a coincident capillary hemangioma.
These lesions, when occurring around the eye and orbit, have led to challenging management issues. Balancing systemic and local steroids with significant side effects has meant that lesions may not have been undertreated, resulting in slower resolution or residual lesions.
Systemic Beta blockers have been the mainstay of this therapy since it’s discovery. Now topical Beta blockers are also proving to be effective. The use of topical Beta Blockers further reduces systemic side effects while still being effective in accelerating the regression of the capillary hemangioma.
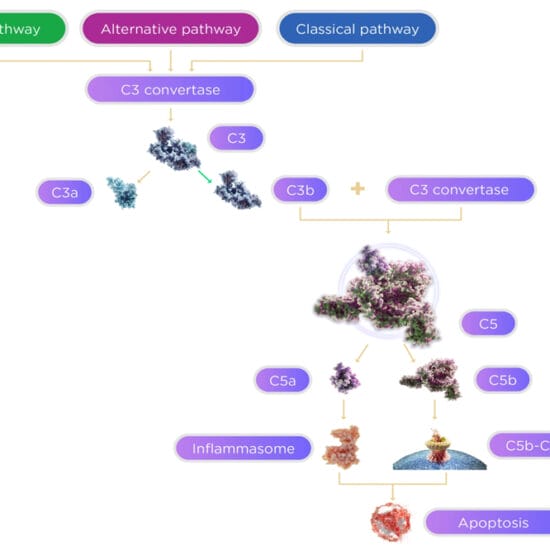
Retinal Gene Therapy for Choroideremia
Choroideremia, caused by mutations in the gene CHM, is characterised by atrophy of the choriocapillaris and retinal pigment epithelium. It can lead to progressive loss of the visual field with visual acuity (VA) remaining close to normal until the disease reaches its latter stage.5
A study reported early improvement in visual acuity in two out of six patients treated with retinal gene therapy for the disease, however noted that in some patients the effect of preserving VA with retinal gene therapy may take several years to become apparent.5
Retinal gene therapy was delivered in the form of non-mutated CHM in an AAV vector by subfoeval injection into the area of the retinal pigment epithelium and photoreceptor cells of one eye in each patient. Untreated (control) eyes of each patient were the eyes with better VA at baseline and these eyes did not receive treatment.5
The study reported early visual acuity improvement experienced by the two patients was sustained at 3.5 years after treatment, despite progressive degeneration in each of the control eyes. The remaining four patients experienced differing results. VA for one patient who developed a cataract at two years requiring removal, recorded visual acuity at 3.5 years similar to the level recorded 12 months after surgery and before the cataract was clinically significant. A patient who received a lower vector treatment dose than other patients declined in visual acuity at 3.5 years while the remaining two patients maintained vision in their study eyes that was close to baseline while VA in their control eyes declined.5
Comment
Gene therapy has heralded the promise for improving vision in patients with inherited eye disease where currently there is no therapy and the prospects are for a life time of impaired vision. Current techniques enable a single gene to be replaced. The challenges in the technique include developing a method to deliver the normal gene to the appropriate site in the eye. This is accomplished by using a modified viral vector to carry the ‘payload’ gene to the site. Much work is required to optimise the viral vectors to enhance the delivery of the gene product to the site. Current technologies means the gene therapy needs to be delivered to the subretinal space to maximise the transfer of genes to the affected cells.
The choroideremia trial reported this year by Thomas et al builds on the RPE65 gene therapy trials. These trials highlight the success of the technique in improving vision but remind us that the improvements are incremental and not the dramatic ‘cure’ that we all hope for our patients. The gene therapy trials also emphasise the challenges in monitoring visual function in individuals with extremely low vision and developing assessments that enable changes to be monitored to see the effectiveness of therapies.
Recent advances in the application of genomics to paediatric eye care emphasises the importance of regular monitoring for patients with these conditions. This will assist in monitoring disease progression, but also importantly, to educate the families about treatment advances.
Associate Professor John Grigg MBBS (QLD) MD (SYD) FRANZCO FRACS is Associate Professor and Head, Discipline of Ophthalmology Save Sight Institute, Sydney Eye Hospital Campus Sydney Medical School, The University of Sydney. He is also a Consultant Ophthalmologist at Sydney Eye Hospital and an Honorary Visiting Medical Officer at The Children’s Hospital Westmead. A/Prof. Grigg has sub-specialties in glaucoma, cataract, paediatric and genetic ophthalmology and clinical electrophysiology. He completed his training at Sydney Eye Hospital and undertook fellowships in Australia and the UK. His research interests are genetic eye disease, glaucoma management and electrophysiology of the visual system.