Patients with profound vision loss may one day regain some level of sight thanks to research exploring ways to deliver optogenetic proteins to the retina.

Dr Dario Protti
It is still common to hear patients with inherited retinal degeneration say they have been told “nothing can be done”. Even before the advent of new approaches, such as gene therapy, this was not true: many things could be done to assist patients in their activities of daily living. However, active ‘treatments’ are indeed limited.
One of these is a gene therapy (voretigene neparvovec, or Luxturna) which has been approved in Australia to treat a form of early-onset retinal degeneration (Leber Congenital Amaurosis Type II; LCA2).1
In November 2020, the first procedure to surgically deliver Luxturna to a teenager with LCA2 in Australia was undertaken by one of the authors (Assoc/Prof Matthew P. Simunovic) at the Children’s Hospital Westmead. This treatment is approved by the Therapeutic Goods Administration for patients with bi-allelic mutations of the RPE65 gene. As anticipated from the results of the Phase III trials, treatment has had a profound effect on these patients’ ability to perform activities of daily living (e.g. they are able to navigate under street lighting at night without a cane for the first time in their life). However, Luxturna is only suitable for one in 300,000 individuals at most (and possibly as few as one in 1.2 million).2 More than 300 genes and loci have been identified as causing inherited retinal degeneration.3 Furthermore, a firm genetic diagnosis cannot be established in 30% of patients attending specialised clinics.4 Finally, current gene therapy approaches are unsuitable for people with advanced disease.3 This is because current gene therapy aims to prevent, or slow, the degenerative process.

Dr Lay Khoon Too
This leaves a significant and unmet need for approaches that are independent of genetic cause, and which are suitable for people with advanced loss of sight.3
Electronic retinal implants were once heralded as offering true hope for such patients. However, neither of the two devices which achieved regulatory approval in the USA/EU (the Argus II and the Alpha IMS) are currently produced (although there are ongoing efforts to advance this approach).
The final common pathway to vision loss in inherited and age-related retinal degenerations is loss of the photoreceptors. However, the light-insensitive second-order (bipolar cells) and third-order (retinal ganglion cells) neurones that relay information from the rods and cones to the brain persist, even in advanced disease.5
WHAT IS OPTOGENETICS?
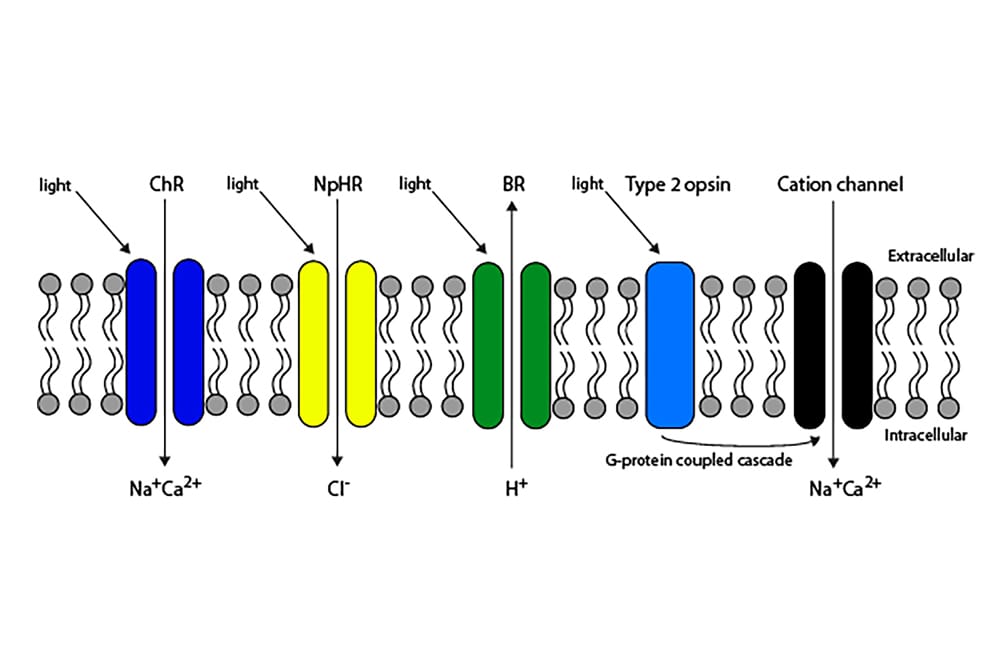
In its broadest sense, optogenetics refers to any approach in which new genetic information encoding a light-sensitive protein is introduced into neurons with the purpose of stimulating them with light.5 The technique was initially conceived as a tool for precisely targeting and studying the role of individual neurons in specific neural circuits.6 In the context of vision restoration, optogenetics primarily aims to restore vision by making second- and/ or third-order neurons light-sensitive.5 This is achieved through the introduction of coding sequences via gene therapy for two broad types of light-sensitive protein: Type I (microbial) or Type II (mammalian) opsins (either of which can be modified; see Figure 1). Adeno-associated viral vectors are, at present, the primary treatment vector used; these do not cause human disease in the natural form, provide stable protein production following treatment, and minimise the potential for inflammation.5

Associate Professor Matthew P. Simunovic
The earliest experiments involving optogenetic gene therapy for vision restoration utilised channelrhodopsin. Channelrhodopsins are the only known ion channels gated by light.7 The chromophore – all-trans-retinal – remains covalently attached to the protein component following light absorption (photoisomerising to 13-cisretinal), thus preventing ‘bleaching’, which is a feature of Type II opsins.5 In contrast to the latter, a series of steps is, therefore, not required to store the channelrhodopsin to its light-sensitive state. Channelrhodopsins used in early optogenetics typically had peak sensitivities at short visible wavelengths and (in common with all Type I opsins) required a high rate of quantal hits to produce an action potential when expressed in neurons. These facts have several important ramifications concerning their use in vision restoration. First, blue light is generally less abundant than longer-wavelength light in low luminance conditions encountered in the natural environment.8 Second, the ocular media demonstrate significant absorption of short-wavelength light.9 Third, the operational range of naturally occurring channelrhodopsins is such that the light intensities necessary for activation approach the maximum permission exposure to avoid phototoxicity (n.b. the action spectrum of Type I and Type II phototoxicity peaks at short visible wavelengths).5 More recently, other ‘red-shifted’ Type I opsins have been engineered or identified in organisms;5,10 these avoid many outlined issues by shifting the wavelength of peak sensitivity to longer wavelengths.

Figure 1. Schematic of Type I and Type II opsins. Type I opsins, such as channelrhodopsin (ChR), act by permitting the passage of ions though the cell membrane in response to light. Type II opsins act via a G-protein coupled
second-messenger system, which amplifies the signal.
Human opsins (e.g. rhodopsin, melanopsin) may also be expressed ectopically and appear to operate by commandeering resident G-protein coupled cascades (principally Gi/o/t), which are presumed to enhance sensitivity through signal amplification.5 They have several advantages, including a lower theoretical propensity to elicit an immunological response and improved sensitivity. While they may provide a more extensive operational range, they generally offer inferior kinetics of light response to Type I opsins. In particular, melanopsin suffers from extremely slow kinetics,11 which would limit temporal resolution. A further potential problem with rhodopsin as an optogenetic tool is bleaching/recovery: this is crucially dependent upon the retinal pigment epithelium (RPE) in the normal retina. However, a Müller cell dependent accessory pathway, important in cone photopigment regeneration, may be sufficient when Type II opsins are expressed, for example, in bipolar cells.12 Optogenetic retinal gene therapy offers a significant advantage compared to retinal prostheses in that it may be possible to treat and transfect large areas of the retina, or targeted areas (depending on mode of delivery and vector design).
PROMISING PRE-CLINICAL TRIALS
Numerous pre-clinical trials have demonstrated restoration of vision by optogenetic techniques using a variety of approaches in terms of opsin (Type I versus Type II), method of delivery (intravitreal versus sub-retinal) and vector (typically an adeno-associated virus).5 As a general rule, Type II opsins may offer sensitivities approaching the threshold for cone activation, while Type I opsins offer lower sensitivity by a factor of typically two log units.5 Our lab has previously investigated the ability of a novel Type I opsin, bReaChES to restore high sensitivity vision in a preclinical model of retinal degeneration. Intravitreal therapy with an adeno-associated viral vector containing the sequence for bReaChES results in novel light sensitivity in retinal ganglion cells, which are capable of responding to light at levels consistent with bright indoor lighting and to flicker at rates approaching the upper limits of human temporal resolution.13 Furthermore, bReaChES gene therapy results in restored light sensitivity behaviourally (see Figure 2).13 Our group is currently extending upon these experiments by working on two novel and complementary approaches to optogenetic gene therapy. The first approach is to assess the ability of new Type I and Type II opsin approaches to restore vision in advanced age-related atrophic and inherited forms of macular degeneration (supported by the Macular Disease Foundation Australia). The second approach compares and assesses the efficacy of vision restoration by expression in second-order (i.e. bipolar cell) versus thirdorder (i.e. retinal ganglion cells) neurones. This work is supported by the National Health and Medical Research Council.
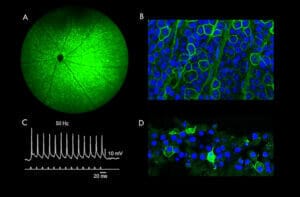
Figure 2. The retina following bReaChES optogenetic gene therapy. (A) Fundus photography displaying expression of the Type I opsin, bReaChES, tagged with a green fluorescent protein. (B) Microscopic honey-combed cellular structure bordered with green fluorescence indicating membranous expression of bReaChES in retinal ganglion cells. (C) Action potentials elicited by bReaChES treated retinal ganglion cells in response to 50Hz light pulses . (D) Microscopic expression of bReaChES in retinal ganglion cells of human retinal explants.
Sahel and colleagues14 have recently offered proof-of-concept for optogenetic approaches for treating advanced sight loss from retinal degeneration. They treated a 58-year-old man who had been diagnosed in his teens with retinitis pigmentosa. He was treated monocularly in his worseseeing eye (vision light perception) with an intravitreal injection of an adenoassociated virus carrying a sequence for a ‘red-shifted’ Type I opsin called ChrimsonR as a part of the Phase I/IIA PIONEER study (ClinicalTrials.gov Identifier: NCT03326336). Because of ChrimsonR’s comparatively poor light sensitivity (over two log units less sensitive than the opsins our group is currently investigating), optogenetic treatment had to be combined with light amplification via stimulus goggles, which provided pulses of 595nm light, which lies close to the wavelength of peak sensitivity for ChrimsonR. The patient was followed-up for 84 weeks following treatment, and their vision was assessed systematically while performing basic vision-guided tasks such as object detection. Additionally, some assessments were performed concurrently with electroencephalography (EEG) to confirm restoration of cortical activity following treatment. The patient only commenced training with the stimulus goggles 4.5 months post-treatment but did not report any subjective benefit for seven months of training (i.e. 11.5 months post-treatment). However, the patient demonstrated behaviour consistent with the ability to recognise the presence of an object on a table and to identify and grasp tumblers. Furthermore, changes on the EEG were consistent with stimulation of the visual cortex with the goggles activated. Although these results are preliminary, they offer the first suggestion that optogenetic proteins delivered to the retina of blind patients have the ability to restore some level of vision in patients with profound vision loss.
Matthew Simunovic MB BChir PhD FRANZCO is an Associate Professor of Ophthalmology at the Save Sight Institute where he leads the Retinal Disease and Rescue Group. Additionally, he is a consultant ophthalmic surgeon at Sydney Eye Hospital and the Sydney Children’s Hospitals Network where he subspecialises in surgery and diseases of the retina. Assoc/Prof Simunovic’s research interests include emerging treatments for retinal diseases, experimental vitreoretinal surgery and visual psychophysics. He was the first Australian ophthalmologist to be awarded a Career Development Award from the Foundation Fighting Blindness USA and is supported by major grant funding from the Macular Disease Foundation Australia and the NHMRC.
Dr Dario Protti PhD is a Senior Lecturer, Neuroscience Theme, School of Medical Sciences, University of Sydney. Dr Protti’s research focuses on the identification of the synaptic networks involved in the extraction of specific features of the visual world. In the last 20 years, he contributed to our current understanding of the physiological mechanisms involved in signal transmission in the retina as well as to the identification of membrane channels, neurotransmitters and receptors mediating processes such as contrast detection, gain control and synaptic transmission. His areas of expertise include every single technique employed to study retinal function at the cellular level: voltage-, current‑ and dynamic-clamp recordings, optogenetics, calcium and glutamate imaging, and multielectrode array recordings.
Dr Lay Khoon Too PhD is a senior research scientist of Retinal Disease and Rescue Group at the Save Sight Institute, University of Sydney. Her research explores optogenetic gene therapy and cell-based approaches to restore vision loss caused by retinal diseases.
References
- Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65- mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390(10097):849-60.
- Chao DL, Burr A, Pennesi M. RPE65-Related Leber Congenital Amaurosis / Early-Onset Severe Retinal Dystrophy. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews((R)). Seattle (WA)2019.
- Simunovic MP, Mack HG, Ayton LN, Hassall MM. Gene Therapy, Diet and Drug Approaches to Treating Inherited Retinal Disease. Reference Module in Biomedical Sciences: Elsevier; 2021.
- Moore AT. Genetic Testing for Inherited Retinal Disease. Ophthalmology. 2017;124(9):1254-5.
- Simunovic MP, Shen W, Lin JY, Protti DA, Lisowski L, Gillies MC. Optogenetic approaches to vision restoration. Exp Eye Res. 2019;178:15-26.
- Deisseroth K. Optogenetics: 10 years of microbial opsins in neuroscience. Nat Neurosci. 2015;18(9):1213-25.
- Lin JY. A user’s guide to channelrhodopsin variants: features, limitations and future developments. Exp Physiol. 2011;96(1):19-25.
- Wyszecki G, Stiles WS. Color science : concepts and methods, quantitative data and formulae. New York; Chichester: John Wiley & Sons; 2000.
- Norren DV, Vos JJ. Spectral transmission of the human ocular media. Vision Res. 1974;14(11):1237-44.
- Marshel JH, Kim YS, Machado TA, Quirin S, Benson B, Kadmon J, et al. Cortical layer-specific critical dynamics triggering perception. Science. 2019;365(6453).
- De Silva SR, Barnard AR, Hughes S, Tam SKE, Martin C, Singh MS, et al. Long-term restoration of visual function in end-stage retinal degeneration using subretinal human melanopsin gene therapy. Proc Natl Acad Sci U S A. 2017;114(42):11211-6.
- Wang JS, Kefalov VJ. The cone-specific visual cycle. Prog Retin Eye Res. 2011;30(2):115-28.
- Simunovic MP, Shen W, Kilpatrick D, Protti DA, Leamey C, Too LK, et al. Optogenetic restoration of vision with bReaChES. Clin Exp Ophthalmol. 2019;47:61.
- Sahel JA, Boulanger-Scemama E, Pagot C, Arleo A, Galluppi F, Martel JN, et al. Partial recovery of visual function in a blind patient after optogenetic therapy. Nat Med. 2021;27(7):1223-9.